Luteal Phase Deficiency: Pathophysiology, Diagnosis, and Treatment
Authors
INTRODUCTION
“When a thing ceases to be a subject of controversy, it ceases to be a subject of interest.”
William Hazlitt (1778–1830), English essayist
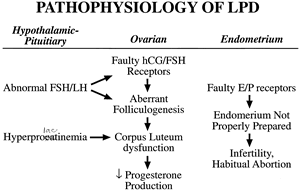
In an era of ever-increasing knowledge of the symphony of physiologic events surrounding ovulation, fertilization, and implantation of human oocytes and embryos, born out of previously unimaginable technologic advancement, few scientific puzzles have been explored more times than the entity synonymously known as luteal phase insufficiency, inadequacy, defect, or deficiency. This chapter uses the term luteal phase deficiency (LPD). Although it successfully can be argued that LPD is the most common abnormality of the menstrual cycle, its significance as a disease entity is shrouded in controversy over its incidence, means of diagnosis, and legitimacy of treatment. Since the first formal description of LPD in 1949 as a possible cause of infertility and recurrent miscarriage by Jones,1 innumerable investigations have been undertaken in an effort to verify its existence or to characterize its pathophysiology, diagnosis, and treatment. The consensus of the literature is that LPD does exist and that its cause is multifactorial (e.g., abnormal folliculogenesis, inadequate luteinizing hormone [LH] surge, inadequate secretion of progesterone by the corpus luteum, aberrant end-organ response by the endometrium). These possible causes are not mutually exclusive in individual patients with LPD, which adds more fuel to the controversy. Obscuring the issue further are related syndromes, such as hyperprolactinemia and hypothyroidism, that are associated with LPD and may induce luteal phase abnormalities secondarily in the presence of otherwise normal ovarian and endometrial function.
The most problematic aspect of a review of LPD is the persistent lack of universal standards for definition and diagnosis. Jones1 claimed that the most accurate means of diagnosis was performing daily assays of serum progesterone throughout the luteal phase but noted that this is impractical except in a research setting. Divergent opinions persist regarding the clinical gold standard for measurement of LPD; this has led to tremendous variability in the estimated prevalence and cure rates in infertile populations. This chapter reviews seminal points regarding LPD that have been reported in a wide variety of investigations.
DEFINITION
The original definition of LPD presented by Jones in 19491 was a corpus luteum defective in progesterone secretion, which in turn was a cause of infertility or early spontaneous abortion. Further investigation led to a broadening of this definition to include a short luteal phase interval (<12 days between ovulation and menses) with relatively normal progesterone concentrations, a normal-length luteal phase with inadequate progesterone production, or inadequate endometrial response to otherwise normal progesterone concentrations.2 Because of the possibility of sporadic abnormalities of luteal function in otherwise fertile women, most experts agree that the diagnosis of LPD can be made only after repeated testing. LPD is relevant in the clinical setting only if it is present in most menstrual cycles in a patient. The requirement for two consecutive menstrual cycles, as currently applied to the diagnosis of LPD, is arbitrary.
PREVALENCE AND INCIDENCE
An accurate estimation of the true prevalence of a disease requires an understanding of the sensitivity of diagnostic modalities and a definition of the abnormality. In the case of LPD, in which the diagnosis has been based variably on single or multiple serum progesterone measurements, urinary progesterone metabolites, salivary progesterone levels, and endometrial histology, the true prevalence is in dispute. Estimating the incidence of LPD in a normal, fertile population is extremely difficult because of the apparently sporadic nature of the disorder. Much of the difficulty centers around the circular pattern of verification of one test by comparison with another unproven test with inherent and immeasurable bias. Also, there is a lack of uniformity of the standards for specific tests used for diagnosis (e.g., endometrial biopsy specimen out-of-phase by≥2 days versus≥3 days).
Despite these obstacles, several reports in the medical literature have attempted to provide estimates of prevalence. Jones,3 in a review of clinical experience, reported LPD diagnosed by out-of-phase (≥3 days) endometrial biopsy specimen in 3.5% of infertile patients and 35% of cases of recurrent miscarriage. Using similar methods, Balasch and Vanrell4 found that 13.5% of infertile patients had evidence of histologically diagnosed LPD, with an incidence of 32.5% for recurrent miscarriage. The latter authors noted that these results were supported by findings of other studies,5,6 despite failing to account for differences in the definition of an out-of-phase biopsy specimen.
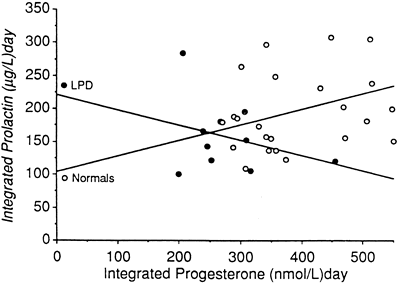
Investigation into the exact incidence of LPD in the general population has proved to be equally elusive. Li and colleagues,7 in a study of 227 menstrual cycles, reported an incidence of out-of-phase (≥3 days) biopsy specimens in 4.4% of fertile controls compared with a 14% incidence in their infertile population. A study of 104 otherwise normal, ovulatory women undergoing tubal anastomosis or donor insemination revealed no cases of retarded endometrium on biopsy, however.8 Another study of 10 sequential monthly endometrial biopsy specimens in five fertile women showed an incidence of sporadic and sequential out-of-phase (≥3 days) biopsy specimens of 31.4% and 6.6%, similar to those reported for an infertile population.9 These findings confirm other reports by Balasch and colleagues,10 Grunfeld and coworkers,11 and Peters and associates12 that there is no difference in the sporadic rate of appearance of retarded endometrial biopsy cycles between fertile and infertile groups, questioning whether testing for LPD in an infertile population is identifying a real disease or a normal variant of the menstrual cycle. The incidence of LPD also may be influenced by the presence of mitigating factors, such as dieting,13 recent childbirth,14 lactation,15 extremes of reproductive age,16,17,18 exercise,19 endometriosis,7,20 unexplained infertility,7 and hyperprolactinemia (Fig. 1).21,22,23,24
|

This lack of a consensus was addressed by a 1991 symposium of international experts, with the following two generalized conclusions regarding the incidence of LPD:25 First, LPD occurs in 3% to 10% of infertile patients presenting to a subspecialty referral practice. Second, although normal women sporadically have deficient luteal production of progesterone, infertile women with LPD experience it in most of their spontaneous menstrual cycles.
NORMAL PHYSIOLOGY OF THE CORPUS LUTEUM
Normal corpus luteum formation and function begin in the follicular phase with recruitment of a cohort of growing follicles primarily under the influence of follicle-stimulating hormone (FSH). By cycle day 7, usually a single dominant follicle emerges as the major source of circulating estradiol (Fig. 2). The microstructure of this dominant follicle comprises an inner layer of granulosa cells, whose aromatase activity converts ovarian androgens to estrogens. The androgens that originate in the outer thecal layer of cells diffuse into the inner granulosa layer. With the estradiol peak and the LH surge at mid cycle, angiogenic factors, such as vascular endothelial growth factor, cause extension of blood vessels past the defunct basement membrane into the previously protected granulosa layer, providing cholesterol substrate attached to low-density lipoproteins (LDLs) for progesterone production (Fig. 3).26 The dominant follicle is transformed into the corpus luteum. By this point in the cycle, granulosa cells that previously produced only low levels of progesterone have acquired LH receptors under the influence of FSH. In the luteal phase, under the stimulatory influence of pulsatile LH in nonconceptive cycles or human chorionic gonadotropin (hCG) from the early embryo and trophoblast in conceptive cycles, the corpus luteum continues to produce and secrete progesterone and other hormones (e.g., relaxin, inhibin, estradiol, oxytocin, prostaglandins).
|

|
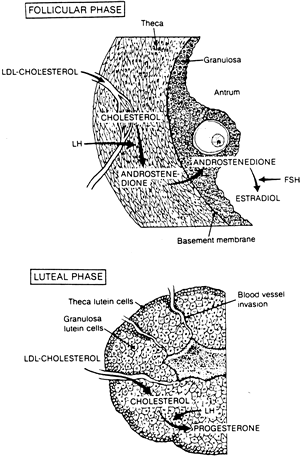
Two distinct steroidogenic cell types have been identified within the human corpus luteum: large luteal cells derived from the granulosa cells (granulosa lutein cells), which produce large quantities of progesterone but are not LH receptive and account for basal progesterone secretion, and small luteal cells derived from thecal cells (theca lutein cells), which are LH receptive in the second half of the luteal phase and are responsible for pulsatile changes in circulating progesterone levels (Fig. 4).27,28,29 During the first half of the luteal phase, increased amounts of estradiol and progesterone are secreted by the corpus luteum with no temporal correlation to LH pulses, primarily by the large granulosa lutein cells. During the second half of the luteal phase, estradiol and progesterone levels in the blood fluctuate in phase with LH pulses, presumably as a result of intermittent secretion by the small lutein cell population (Fig. 5).30,31,32 Steroid secretion by each cell type is affected by complex paracrine interactions mediated by peptide hormones produced by the large luteal cells. Small cells are virtually devoid of synthetic capacity for peptidergic compounds.31,33
|

|

The best characterized of these modifying peptides is oxytocin. Elaborate microdialysis studies of intact human corpus luteum have shown that oxytocin, stimulated by prostaglandin F2α from the uterus, exhibits a stimulatory effect on progesterone secretion in the first half of the cycle via local estradiol mediation.34,35 As the luteal phase progresses, the stimulatory effects of oxytocin and prostaglandin F2α on estradiol decrease, and luteolytic factors, such as tumor necrosis factor from luteal cells and invading macrophages, play a more active role in disruption of corpus luteum function.35 In the absence of blastocyst implantation and with no hCG secretion, there are diminished levels of circulating progesterone, causing inadequate endometrial support and eventual sloughing at menses.
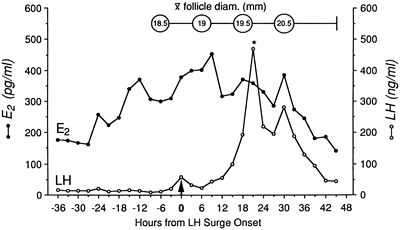
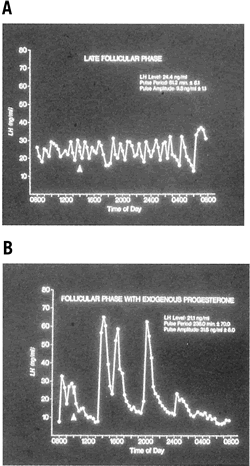
The preparation of the endometrium for implantation of the blastocyst is the end result of a well-orchestrated series of endocrine, paracrine, and autocrine events that affect the synthesis of hormones and their receptors in the hypothalamic-pituitary-ovarian axis. Early in the follicular phase, FSH, with estradiol, acts to induce its own receptors and those of LH on the maturing granulosa cells. FSH also serves to induce aromatase activity and increase production of inhibin from growing follicles. Estradiol induces transcription of mRNA for production of its own receptors and for progesterone receptors in the developing endometrium. Increased estradiol production at mid cycle creates positive central feedback on LH secretion, leading to a surge. The LH surge is characterized by a rapid ascending phase lasting 14 to 17 hours and a more gradual descending phase, with a total duration of 48 to 54 hours (Fig. 6).36,37 The slow decline in mid cycle serum LH levels has been hypothesized as a mechanism to promote early progesterone production and corpus luteum support.3
|
An adequate balance of endometrial proliferation stimulated by estradiol with secretory transformation resulting from progesterone leads to the purported window of implantation between days 20 and 24 of the classic 28-day menstrual cycle. The implantation window is characterized histologically by expression of cell surface adhesion proteins known as integrins, specifically, the avβ3 integrin.38 Although the exact role of integrins (if any) in implantation is unknown, they serve as consistent cell markers of receptivity. Differential expression of endometrial estradiol and progesterone receptors throughout the normal menstrual cycle allows for an appropriate end-organ response, while maintaining the balanced influence of these two hormones. Although the corpus luteum produces a variety of hormones, the ability of estradiol and progesterone replacement alone to create a successful, receptive endometrial environment for donor egg in vitro fertilization (IVF) recipients who are down-regulated with gonadotropin-releasing hormone (GnRH) agonists emphasizes the importance of these two steroids over other corpus luteum products. The rest of this chapter focuses on luteal dysfunction as an abnormality of progesterone secretion and effect, with the bulk of scientific evidence presented focused on this area.
PATHOPHYSIOLOGY
LPD is a heterogeneous disorder that has been shown and investigated in several primate species besides humans. As previously noted, the lack of a diagnostic gold standard has led to varying definitions and myriad hypotheses as to the underlying defect within the menstrual cycle. There is good evidence for the existence of several different causes for LPD, one or more of which may exist in the same woman. In general, the proposed pathophysiologic mechanisms may be divided into three categories centered around the corpus luteum as the primary functional unit: preparation, production, and response (Fig. 7). The basic tenets of each are discussed.
|
Preparation: Inadequate Folliculogenesis
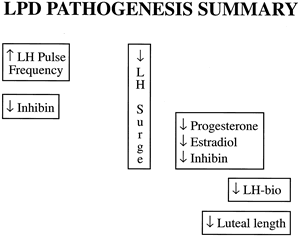
Because the corpus luteum develops from its predecessor, the dominant follicle, it is logical to assume that any defect in the formation or function of the latter could lead to LPD. Early investigation in nonhuman primates by DiZerega and Hodgen39 and Stouffer and Hodgen40 showed that diminished FSH secretion in the early follicular phase induced by administration of porcine follicular fluid rich in inhibin led to inadequate luteal progesterone secretion. They showed that inadequate follicular phase levels of FSH and estradiol ultimately led to incomplete differentiation of luteal cells that were less sensitive to LH and hCG stimulation. Wilks and colleagues41 previously described a group of rhesus macaques who had spontaneous LPD (decreased progesterone levels) and decreased follicular phase FSH levels. Human studies also showed lower follicular FSH levels or diminished FSH-to-LH ratios in women with LPD diagnosed by serum progesterone or histologic criteria.42,43,44,45,46 The exact cause for diminished FSH secretion or effect in spontaneous menstrual cycles with LPD is unknown, but there has been speculation that this finding is due to inhibin or inhibin-like substances that either suppress pituitary FSH secretion or alter its bioactivity. An investigation by Molskness and colleagues47 showed diminished bioactive FSH and estradiol secretion, increased bioactive LH secretion, and lengthening of the follicular phase in rhesus monkeys treated with human inhibin A early in their menstrual cycles. These animals experienced decreased mean luteal progesterone levels despite normal luteal phase length and normal levels and patterns of luteal LH secretion compared with controls. A similar association between a prolonged follicular phase and subsequent LPD has been noted in spontaneous human cycles.11,48
Despite this compelling evidence that abnormalities of follicular phase FSH lead to LPD, other investigations using more frequent sampling techniques in spontaneous cycles with out-of-phase biopsy specimens and diminished integrated luteal progesterone levels showed no differences in follicular phase FSH (immuno- and bioactive) and estradiol levels compared with controls.11,49 To confuse the situation further, and in contrast to the animal studies previously noted, lower levels of inhibin were detected in the early follicular and luteal phases of cycles in women with LPD.49,50 Altogether, we currently believe that abnormalities in folliculogenesis with or without FSH deficiencies can cause LPD, but this mechanism is not the common pathogenesis for LPD as found in most women.
Although some investigators have implicated follicular phase FSH as a crucial factor in later corpus luteum function, others have focused on follicular phase LH—specifically, its pattern of pulsatile secretion early in the menstrual cycle. Soules and coworkers51 first reported abnormal pulsatile patterns of LH secretion in four fertile women with histologically diagnosed LPD. They showed an increased early follicular LH pulse frequency compared with control women. In a larger and more definitive study,52 these investigators showed that women with LPD maintain a fixed pattern of increased pulsatile secretion throughout the follicular phase, in contrast to normal women, who experience an acceleration in pulse frequency with the approach of ovulation. This pattern emerged despite similar follicular phase peak and integrated estradiol levels and no change in follicular phase mean LH and FSH (immuno- and bio-levels).49 They speculated that the increased LH pulse frequency may result from lower serum progesterone levels in the preceding luteal phase because progesterone has been shown to slow LH secretion by the pituitary.51 Soules and coworkers53 tested this latter hypothesis in a study of six normal women with LPD induced by administration of a GnRH antagonist in the midluteal phase. Analysis of LH pulses in the early follicular phase of the ensuing menstrual cycle showed no increased frequency over baseline, however, suggesting that the intrinsic defect lay within the neuroendocrine control of the GnRH pulse generator and not in the antecedent corpus luteum. These findings were confirmed by a similar pulse analysis study by Suh and Betz.54
The immediate sequela of an increased LH pulse frequency in the first half of the menstrual cycle seems to be diminished levels of bioactive LH in the luteal phase, with concomitant decreases in progesterone pulse amplitude and mean serum progesterone levels (Fig. 8).49,52 These observations in spontaneous menstrual cycles confirmed an earlier study in which luteal bioactive LH was diminished after a supraphysiologic gonadotropin pulse frequency was induced in the follicular phase in normal women.55 In a study of a group of young women with exercise-related and diet-related LPD, analysis of early follicular LH pulses showed slower frequencies compared with normal women.56 This seeming contradiction may serve only to show how perturbation of the gonadotropin pulse generator in one direction or another during the follicular phase can alter subsequent corpus luteum function.
|
Whether inhibin is part of the pathophysiology of LPD is uncertain. As previously noted, early and mid follicular levels of inhibin have been noted to be diminished in LPD cycles despite normal levels of FSH and estradiol.49,50 Soules and coworkers49 speculated that lower levels of inhibin may reflect a differential insensitivity to FSH that does not affect estradiol secretion. The difficulty in proving such a hypothesis was the significant amount of overlap between inhibin levels in normal and LPD women. In addition, the relative lack of specificity of the inhibin assay available at the time of that analysis did not allow accurate quantification or identification of the active dimeric forms of inhibin (A and B). A more recent study by Molskness and colleagues,47 in which supraphysiologic levels of inhibin A were given to monkeys to induce LPD, further clouds the issue of inhibin. It would be interesting to know whether there are any changes in circulating levels of inhibin A or B during the menstrual cycles of women with spontaneous LPD.
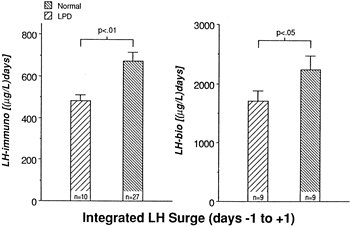
Studies of ovulation induction in which ovulation was triggered using small amounts of hCG or LH by Jones and Madrigal-Castro57 and Van de Wiele and coworkers58 first raised the possibility of mid cycle LH surge inadequacy as a potential mechanism for LPD. These investigators showed that an inadequate surge duration led to aberrant luteal function. Later investigations using daily blood sampling confirmed that LPD cycles were characterized by attenuated integrated LH levels around the time of the mid cycle surge (Fig. 9).49,54 The authors hypothesized that the more frequent but stunted LH pulses throughout the follicular phase may down-regulate LH secretion at mid cycle, leading to a blunted LH surge.54 At present, there are no studies to prove or disprove this theory. These findings in LPD cycles are difficult to explain, considering a lack of correlation between integrated LH surge levels and progesterone production in normal menstrual cycles.59 Taken together, one might hypothesize that there is a minimal threshold for an LH surge that ensures, but does not control, the amount of progesterone subsequently secreted.
|
Other investigations of LPD cycles have focused on the development and function of the preovulatory (dominant) follicle. Check and colleagues60 evaluated women with histologically proven LPD sonographically and found that they ovulated more frequently from relatively small dominant follicles (<17 mm mean diameter). This finding was confirmed by Ying and associates,61 who noted that 39% of LPD cycles were characterized by small dominant follicles compared with those of normal women at their institution. They also found that this percentage decreased to 6% in histologically corrected LPD cycles. These findings were later refuted, however, by other investigations of spontaneous LPD cycles during which daily ultrasound monitoring of follicles was performed.11,49
Production: Inadequate Progesterone
The endocrine function of the corpus luteum, as is the case for other endocrine organs, depends on the interplay of multiple factors: (1) Sufficient precursor molecules must be available for eventual conversion to the active hormone; (2) tropic stimuli must be present in the proper quantity and quality to promote hormone synthesis; (3) the target cells of the tropic stimuli must have the requisite mechanism for receiving and converting that message for intracellular response; (4) the actual cellular production of hormone must be adequate; and (5) the activity of the hormone secreted must be sufficient.
The precursor for biosynthesis of progesterone is plasma cholesterol transported as LDL. According to the model presented by Carr and colleagues,26 the poorly vascularized granulosa cells in the follicular phase normally are isolated from plasma LDL cholesterol (see Fig. 3). After ovulation, as the microvascular network in the thecal layers invades the luteinized granulosa cells, local LDL concentrations approach plasma levels, providing the substrate for progesterone production. In support of this substrate theory, observers of corpus luteum anatomy have found maximal capillary enlargement or dilaton to have occurred by luteal day 7 or 8, the time of maximal progesterone secretion.62,63 Conceivably, inadequate vascularization could undermine the process of progesterone production, as has been noted histologically in several case studies.64,65 Alternatively, short supply or abnormal forms of LDL cholesterol could lead to inadequate corpus luteum output. Illingworth and colleagues66 reported that women with abetalipoproteinemia, a syndrome marked by extremely low amounts of circulating LDL, have very low serum levels of progesterone (<2 ng/mL) despite otherwise normal menstrual cycle parameters. Serum levels of LDL decrease during the luteal phase in normal menstrual cycles67; however, no difference in luteal phase serum lipoprotein levels between women with normal and LPD cycles was found in an investigation by Hansen and colleagues.68
Progesterone is secreted in a pulsatile fashion by the corpus luteum in normal and LPD women in response to LH secretion by the pituitary (Fig. 10).31,52,59 Intuitively, disruption of LH secretion could lead secondarily to deficiencies in progesterone production by the corpus luteum. Soules and coworkers52 studied luteal phase gonadotropin secretion during LPD cycles and showed that there was no significant difference in LH pulse frequency or amplitude compared with controls. The women studied experienced normal slowing of pulse frequency and an increase in pulse amplitude during the transition from follicular to luteal phases. This slowing of the LH secretion pattern previously was shown to be controlled by progesterone feedback on the GnRH pulse generator, mediated via endogenous opioid peptides (Fig. 11).51 Despite apparently normal patterns of luteal phase LH secretion, bioactive levels of LH were decreased significantly 6 to 11 days after a mid cycle surge, suggesting that there is a qualitative defect in the stimulus to luteal cells.49
|

|
In a similar fashion in conceptive cycles, continuation of pregnancy depends on tropic support of the corpus luteum by hCG elaborated by the early trophoblast. Apparently the corpus luteum becomes increasingly less sensitive to LH stimulation with a preferential response to hCG support as ovulation becomes more remote. Without adequate hCG, luteolysis takes place, with ensuing pregnancy failure. Conceivably, alterations in the hCG molecule affecting its bioactivity or in the pattern of its secretion could disturb this luteal rescue function. This hypothesis is theoretical at present but offers a provocative target for future research.
The aforementioned two-cell compartmentalization of the corpus luteum has led to studies of LPD based on hypothetical failure of specific cell types. Hinney and colleagues28 investigated LH pulsatility in 38 women with LPD diagnosed by two consecutive cycles marked by diminished mid luteal progesterone levels. They described three distinct patterns of luteal phase failure. Group 1 consisted of 16 women (42%) who had no LH secretory episodes and had significantly lower basal LH levels compared with 14 control subjects. Group 2 consisted of 13 women (34%) who had normal LH secretory episodes despite low estradiol and progesterone concentrations that did not respond to the LH pulses. Group 3 consisted of nine women (24%) who had normal LH pulses with an adequate progesterone response but inadequate basal secretion of progesterone. All immunoreactive LH pulses also were found to be bioactive. Group 1 subjects were designated as having a hypothalamic cause of LPD (inadequate stimulation). Group 2 women, because of their lack of response to LH pulses, were categorized as having a small cell defect. Group 3 women, with low basal levels of progesterone and appropriate responses to LH episodes, were categorized as having a large cell defect. These findings led to intriguing implications for the management of patients with LPD. Patients found to have an increase in serum progesterone levels in response to LH stimulation (large cell defect) may be treated adequately with exogenous hCG; patients with inadequate LH response (small cell defect) may be treated preferentially with progesterone supplementation. Further investigation is needed to verify these findings and to characterize the specific cellular defects within these subtypes.
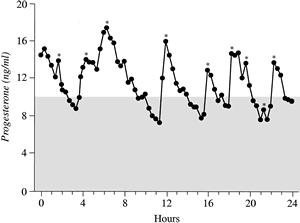
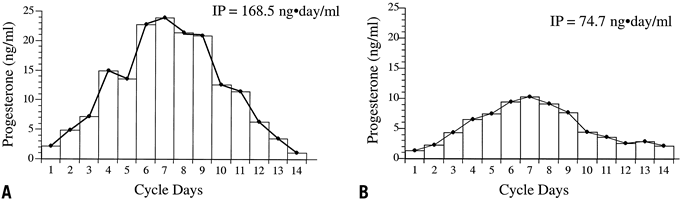
Further analysis of LPD has focused on the quantity and quality of progesterone produced by the corpus luteum. The difficulty in assessing the former is the wide fluctuation in serum progesterone levels over a 24-hour period. One study using frequent blood sampling in normal women during the mid and late luteal phases found the mean percentage variation in progesterone over 24 hours to be 99% and 138% (Fig. 12).59 No identifiable circadian pattern of secretion was established. This level of variation affects the interpretation of single or multiple values in the assessment of luteal adequacy. By increasing the number of data points, however, one may limit the amount of variance and obtain a more accurate estimate of the true mean. This principle, previously referred to as regression toward the mean, allows accurate comparison of women and groups in terms of their corpus luteum secretory capacity using integrated daily luteal phase progesterone levels. Numerous studies that have undertaken such measurements have confirmed that a subset of women exist with decreased quantities of progesterone compared with controls. In some cases, the typical bell-shaped curve of progesterone levels is attenuated, and in others, progesterone peaks early, with significant shortening of the overall luteal phase duration. Intuitively, in cases in which histologic LPD exists despite adequate immunoreactive levels of progesterone in the serum, there may be abnormalities of the bioactive fraction of progesterone. A study by Minassian and Wu70 specifically addressing this issue could not show a difference, however, in the amount of free and protein-bound progesterone between normal and histologic LPD cycles.
|
Response: Effects of Progesterone on the Endometrium
An area of continued interest and controversy in the investigation of LPD pathophysiologic mechanisms is the endometrial response to ovarian steroids. Normally, over the course of the menstrual cycle, the endometrium undergoes dynamic changes in receptivity in response to hormone signals from the ovaries. In general, estradiol and progesterone receptor content increases throughout the proliferative phase, with peak levels in the immediate preovulatory period.71,72 During the secretory phase, under the influence of progesterone, estradiol receptor content declines in all cell types, and progesterone receptors are reduced in glandular epithelium, while remaining constant in stromal and myometrial cells.71 Retention of progesterone receptors in the latter two categories is thought to aid in the maintenance of pregnancy by inhibiting myometrial contractility during gestation. Given these complex and essential functions of progesterone, dependent on the adequacy of its receptor, it has been compelling to address this area as a potential cause of LPD.
The first description of a patient with an abnormal progesterone receptor concentration in endometrium came from Keller and coworkers in 1979.73 In this case report, normal serum levels of estradiol, progesterone, and FSH were documented in the face of multiple abnormal luteal phase endometrial biopsy specimens. Subsequent progesterone supplementation failed to correct the histologic finding of a poorly developed pseudodecidual endometrial reaction. Eventual progesterone binding studies on a sample of the patient’s endometrium revealed normal receptor affinity but a concentration of progesterone receptors only half that of endometrium from control subjects. This case report was a stimulus for investigation and elaboration of steroid receptor changes in the endometrium in women with histologic LPD (Table 1).
TABLE 1. Summary of Estrogen and Progesterone Receptor Concentrations in Endometrial Biopsy Specimens from Women with Luteal Phase Deficiency
No. Subjects | Progesterone Receptors | Estrogen Receptors | ||||
| Study | LPD | Normal | Cytosolic | Nuclear | Cytosolic | Nuclear |
| Gautray (1981)74 | 88 | 79 | Decreased | Decreased | Decreased | Decreased |
| Gravanis (1984)75 | 10 | 7 | NS | Increased | — | — |
| Laatikainen (1983)76 | 14 | 19 | Decreased | — | — | — |
| Levy (1980)77 | 18 | 16 | NS | NS | NS | NS |
| McRae (1984)78 | 14 | 30 | NS | — | NS | — |
| Saracoglu (1985)79 | 20 | 40 | Increased | — | NS | — |
| Spirtos (1985)80 | 10 | 14 | Decreased | |||
LPD, luteal phase deficiency; NS, not significant.
McNeely MJ, Soules MR: The diagnosis of luteal phase deficiency: A critical review. Fertil Steril 50:1, 1988.
Table 1 shows there is little consensus on this issue. All of these studies specifically examined coincidental secretory phase parameters (i.e., serum estradiol and progesterone levels) rather than proliferative phase steroids, which have a more important role in induction of endometrial receptors. As others have suggested,81,82 the absolute number or density of steroid receptors may not be the problem as much as the ratio of estradiol to progesterone receptors in the nucleus and cytosol. In one investigation by Abd-El-Maeboud and coworkers,83 quantification of estradiol and progesterone receptors using a sensitive monoclonal antibody technique in women with histologic LPD revealed significantly lower total progesterone-to-total estradiol receptor ratios compared with other infertile controls. By comparing receptor ratios rather than absolute concentrations between groups, the authors minimized the variability in the latter induced by differing ovarian stimulation regimens. Further investigation of complete menstrual cycles with unbiased morphometric and quantitative evaluation of endometrial steroid receptors is necessary before it can be concluded that there is a unifying endometrial cause for clinical LPD.
From another perspective of a potential endometrial cause, the endometrial response need not be devoid or attenuated but simply delayed. A phase shift could result in suboptimal preparation of the endometrium for attachment and nidation of the blastocyst at the time it enters the uterus. This type of in utero embryonic asynchrony has been well documented in myriad animal species.84 The existence of a window of implantation in the human uterus was established by Hertig and colleagues,85 who examined hysterectomy specimens from women in the luteal phase of their cycles. They discovered that all embryos (n = 8) from hysterectomies performed before day 20 were free-floating, but all embryos (n = 26) from hysterectomies performed after day 20 had implanted. Likewise, several other investigations of the time of maximal uterine receptivity86,87 have placed the window of implantation between postovulatory days 6 and 10 in humans. If the coordinated timing of tubal embryo transport and endometrial receptivity were disrupted, the putative window may be open too late or too briefly for adequate implantation, leading to failed pregnancy or early pregnancy wastage. For this reason, biopsy of the endometrium in the mid to late secretory phase emerged as an attractive bioassay of luteal phase sufficiency. The validity of endometrial biopsy for the diagnosis of LPD and the optimal time to obtain a biopsy specimen remain questionable (see later).
Histologically the mechanism of failure may not depend on steroid hormones and their receptors at all, but rather on the failure of expression of necessary cell surface adhesion molecules (integrins). Lessey and colleagues88 described two types of integrin defects during the mid luteal phases of women with previously unexplained infertility: type I, patients with out-of-phase endometrial biopsy specimens (i.e., LPD) lacking β3 integrin, and type II, patients with in-phase biopsy specimens but still lacking β3 integrin. Even when type I patients were treated adequately, almost one third (5 of 16) failed to express the β3 integrin. Despite correction of a steroid deficiency in the endometrium, a group of women exists who have an endometrium that may be incapable of expressing certain molecules integral to normal implantation and pregnancy maintenance. To date, failed integrin expression in endometrial biopsy specimens has proved to be useful as a diagnostic tool for failed implantation; however, no known, reliable corrective measures exist.89
RELATED SYNDROMES
Hyperprolactinemia
Prolactin is a single-chain polypeptide hormone 198 amino acids in length that circulates in multiple isoforms with varying bioactivity. It is secreted mainly from the anterior pituitary in a pulsatile fashion under tonic inhibition by hypothalamic dopamine. It also is secreted by the placenta and uterine decidua and is thought to play a role in steroidogenesis by luteinized granulosa cells. A landmark study by McNatty and colleagues in 197490 looked at the effects of prolactin on the ability of cultured luteal cells to produce progesterone. They found that neutralization of prolactin in the culture medium with a specific antibody or addition of prolactin over a threshold level led to diminished daily progesterone production in vitro. Further in vivo evidence for modulation of progesterone secretion by prolactin in normal women came from studies of induced mild hyperprolactinemia, in which luteal phase serum levels of progesterone were reduced.21,22 A separate investigation of normal women found decreased luteal progesterone secretion when prolactin levels were decreased to below normal with a dopamine agonist.91 Whether the latter effect was due to direct effects of dopamine on prolactin secretion or on the GnRH pulse generator is unclear, but it was not unexpected given the importance of prolactin for activation of the 3β-hydroxysteroid dehydrogenase enzyme in the ovary.92
Collectively, these studies of normal women with induced changes in prolactin levels support the earlier modulation of corpus luteum steroidogenesis by prolactin as suggested by McNatty and colleagues.90 It seems that optimal corpus luteum function as determined by progesterone secretion depends on a certain level of prolactin, and a change (either increased or decreased) in prolactin leads to decreased levels of progesterone.
Evidence for prolactin’s role as a modulating factor in corpus luteum function has emerged from several studies of women with LPD. Huang and associates23 studied 151 women with out-of-phase endometrial biopsy specimens by measuring serum prolactin levels for 3 to 4 days in the periovulatory period. They found that 33 subjects (22%) had transient elevation in prolactin to greater than 20 ng/mL. They also noted decreased pooled progesterone levels in the mid luteal phase in the transiently hyperprolactinemic women compared with controls. Scaglia and colleagues93 reported nocturnal elevation of prolactin in six women with LPD diagnosed by histologic and serum criteria, four of whom had normal daytime levels of prolactin. Aisaka and coworkers94 found the incidence of transient hyperprolactinemia to be 62.2% in 201 cases of LPD diagnosed by a combination of basal body temperature (BBT) and serum progesterone criteria in their clinic population.
In addition to the transient elevations described previously, sustained hyperprolactinemia may contribute directly to decreased luteal progesterone production. Del Pozo and colleagues24 studied eight women with galactorrhea or hyperprolactinemia with short luteal phases and found integrated luteal progesterone values greater than 2 SDs below control values. These findings corroborate the theory proposed by Bahamondes and associates95 that LPD is part of the natural sequence of events between normal ovulatory cycles and amenorrhea that occurs in women with progressive hyperprolactinemia. Part of the difficulty in interpreting the causal influence of hyperprolactinemia on luteal function is the normal variability of serum prolactin levels. Fujimoto and coworkers96 showed a circadian pattern of prolactin secretion with a definite nocturnal rise in normal women. In addition, secretory prolactin peaks approaching the threshold of normal values (20 ng/mL) regularly appeared in the ovulatory and late luteal periods in their study. Prolactin levels were above normal 20% of the time when drawn between 8 and 10 a.m. in the 28 normal women studied.
A single measurement is of limited value when assessing prolactin secretion as it relates to luteal sufficiency. As an example of the confusion generated with single serum samples, a study by Vanrell and Balasch97 in which prolactin was measured in a single luteal phase blood sample showed a decreased occurrence of abnormal endometrial biopsy specimens for hyperprolactinemic women compared with euprolactinemic subjects.
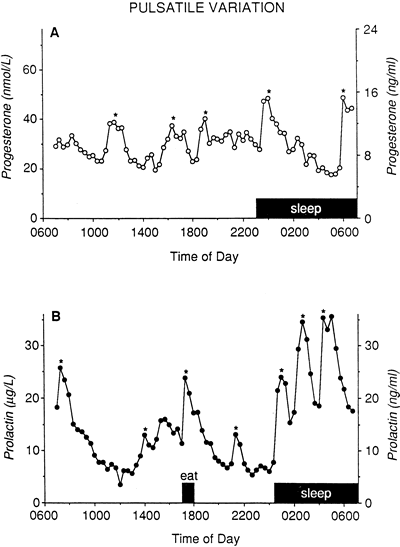
In a further attempt to clarify prolactin’s role in LPD, Soules and associates98 conducted a study of 18 women with histologically diagnosed LPD in which daily blood samples were drawn. They found no correlation between luteal phase integrated progesterone and integrated prolactin levels, and they did not find any differences in the nocturnal prolactin secretion patterns compared with 36 normal control women (Figs. 13 and 14). It would seem that LPD is a common disorder in infertile women with hyperprolactinemia, but the converse is not necessarily true.
|
|
Ovulation Induction
The use of clomiphene citrate (CC) for ovulation induction in women with anovulation or unexplained infertility has been associated with out-of-phase biopsy specimens.99,100,101,102 Luteal phase evaluation from 1964 to 1975 during CC cycles was reported by Garcia and colleagues,101 who noted a 50% incidence of out-of-phase biopsy specimens in infertile patients treated with CC. Cook and colleagues99 found a 50% rate of histologic LPD in otherwise normal women treated with CC and a 73% (16 of 22) rate in previously anovulatory women treated with CC. A similar study of patients with severe oligo-ovulation or anovulation by Keenan and coworkers102 revealed an overall rate of out-of-phase biopsy specimens of 24%, with no significant difference detected between various doses of CC. Bonhoff and associates100 performed a morphometric analysis of midluteal biopsy specimens from patients treated with CC and found increased stromal edema and measurable differences in glandular development compared with fertile controls.
It is unclear why endometrial histologic derangements should arise in otherwise normal, albeit induced, ovulatory cycles. Various theories for CC-induced LPD have been proposed, which vary from increased FSH-to-LH ratios, to inadequate midcycle surges, to relative imbalances of estradiol and progesterone in the luteal phase, to antiestrogenic effects on the endometrium. An investigation by Hsu and coworkers103 using color Doppler ultrasonography of the uterine artery in women with unexplained infertility treated with CC showed decreased uterine perfusion in the early luteal phase, despite a lack of correlation with serum estradiol and progesterone. These findings suggest that a histologic endometrial delay may result from a direct effect of CC rather than an indirect, steroid-mediated effect. Despite this evidence for a causal effect of CC in LPD, in a well-designed study of normal women treated with CC (50- and 150-mg doses) in which each subject served as her own control, only 1 of 16 CC cycles was associated with an out-of-phase biopsy specimen; that 1 cycle had a correspondingly low integrated progesterone level.104 In an attempt to reconcile these reports, it seems that the histologic delay of the endometrium associated with CC is most likely a functional outcome resulting from a relative lack of luteal progesterone with a smaller, direct contribution on the endometrium or its receptors that is as yet incompletely characterized.
The use of gonadotropins for ovulation induction also has been associated with LPD. Incidence reports of 30% have been given for cycles in which human menopausal gonadotropins were used.105 The greatest debate regarding this issue stems from the IVF literature, in which transient decreases in progesterone production have been attributed to follicle puncture.106,107 Evaluation of the luteal phase using serum progesterone levels, BBT charts, and endometrial biopsy specimens after stimulation for IVF has led to a general consensus that LPD occurs 28% to 76% of the time in these cycles.108 Despite this, a meta-analysis of clinical trials using luteal support in IVF cycles by Daya in 1988109 failed to show any significant impact on pregnancy rates compared with controls. More recent studies that examined IVF stimulation that was preceded by down-regulation with a GnRH analogue have shown conclusively, however, that luteal phase support has a positive impact on pregnancy rates.110,111,112,113 Soliman and colleagues114 conducted another meta-analysis of randomized trials of luteal support in 1994 and showed significant improvement in fecundity in IVF cycles that incorporated GnRH agonist down-regulation and hCG or progesterone supplementation after egg retrieval. In their analysis of non-IVF cycles in which human menopausal gonadotropins was used for ovulation induction, no conclusive evidence was available to prove or disprove the efficacy of luteal support.
Exercise
Strenuous exercise has long been recognized as capable of altering menstrual cyclicity. The earliest cross-sectional studies of female athletes showed consistent shortening of the luteal phase, with decreased mid luteal progesterone levels.115,116 Later studies using daily serum, urine, or saliva sampling for calculation of integrated progesterone levels confirmed an increased incidence of LPD with strenuous exercise.117,118,119 Prospective investigations of women with progressive increases in athletic training show a continuum of menstrual irregularities, beginning with progressive shortening of the luteal phase with normal estrogen levels, to oligo-ovulation and anovulation, with eventual hypoestrogenic amenorrhea.120 The ability of exercise to induce LPD or other menstrual disturbances is potentiated when combined with caloric restriction.19 Individual susceptibility is a factor here: Not all women develop LPD or other menstrual cycle abnormalities with the same level of exercise.
In a particularly demonstrative, prospective study by DeSouza and coworkers121 24 women categorized as recreational exercisers running at least 2 hours or 16 km per week were compared with a group of 11 sedentary, eumenorrheic women. Using a duration of less than 10 days or urinary ratio of pregnanediol glucuronide to creatinine of less than 3 μg/mg to define luteal insufficiency, they found that there were no differences in training between normal ovulatory, LPD, and anovulatory runners. They found that 55% of cycles in exercising women were abnormal (LPD or anovulatory) compared with only 10% in the sedentary group. LPD cycles were characterized by a longer follicular phase (17.9 ± 0.7 days) compared with sedentary controls, with a blunted rise in serum FSH levels during the luteal-follicular transition. The authors theorized that this finding, along with a decline in peak LH, represents altered hypothalamic regulation of GnRH and subsequently may account for the observed low estrogen secretion in the early follicular phase. Overall the prevalence of LPD in this group of moderate exercisers was 48%, with an incidence of 79%.
The effects of exercise are mediated in part through opioid compounds in the central nervous system that inhibit the GnRH pulse generator, secondarily slowing LH pulse frequency in the follicular and luteal phases.122,123 Complex mechanisms involving energy balance and circulating levels of the peptide hormone leptin, a product of peripheral adipocytes, may contribute to altered GnRH pulsatility. Although studies to date have confirmed a peak in leptin expression during the luteal phase of the menstrual cycle,124,125 correlations between leptin and progesterone levels have yielded conflicting results.124,126,127,128 Investigation of this relationship specifically in a cohort of women with LPD is lacking.
Recurrent Spontaneous Abortion
Fetal wastage has long been associated with LPD. Women with a history of recurrent spontaneous abortion have been reported to have histologic evidence of LPD in 32.5% to 60% of cases in nonconceptive cycles.4,129 Direct verification of LPD using serum criteria was supplied by Horta and associates130 in an investigation in which serum samples were collected throughout the luteal phase from women with a history of recurrent spontaneous abortion. They showed significantly lower progesterone levels on luteal days 3 to 12 compared with controls. In a study by Babalioglu and colleagues131 in which multiple mid luteal serum progesterone levels and timed endometrial biopsy (TEB) criteria for LPD were used together, women with recurrent spontaneous abortion who had out-of-phase biopsy specimens also had significantly lower progesterone levels. Agreement between these two diagnostic methods in cases of LPD associated with recurrent spontaneous abortion suggests that this subset of patients may have more overt disease that is more readily detectable regardless of test sensitivity.
Other Associations
In addition to the well-recognized associations noted previously, various other entities may contribute to the development of LPD. Dietary caloric restriction may have a profound impact on gonadotropin secretion, secondarily leading to LPD. Anorexia nervosa and bulimia nervosa have been shown to affect luteal function and menstrual cyclicity through various metabolic and endocrine mechanisms. Healthy, normal-weight women also may develop LPD as an intermediate step before amenorrhea during periods of caloric restriction.132 Strict vegetarian diets have a greater association with LPD and menstrual disturbances than do mixed, nonvegetarian diets.133 Endometriosis also has been implicated as a contributing factor in LPD,7,20 but there is no consensus in the literature on this point.134 Thyroid disease, in particular hypothyroidism, also may play a role in LPD via changes in sex hormone–binding globulin and secondary hyperprolactinemia.
DIAGNOSIS
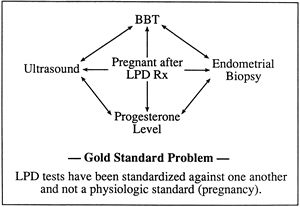
The greatest impediment to complete understanding of the pathophysiology of LPD stems from the lack of established criteria for its diagnosis. Despite the early admonishment of Jones3 that the only way to assess corpus luteum function clearly is by daily measurement of serum progesterone levels, this particular diagnostic method remains a research standard because it is impractical and expensive in the clinical setting (Fig. 15). In an attempt to circumvent the inconvenience of repeated serum measurements, Jones and other investigators turned to the TEB as an end-organ bioassay of luteal hormonal effects. This approach intuitively makes sense because ultimately the major physiologic function of the corpus luteum is to prepare the endometrium for nidation. The problem with the TEB method is that it provides only a “snapshot” of the concert of events leading to implantation that occurred before the time of biopsy. Other investigators chose to measure one or several serum progesterone levels as a subsample of overall corpus luteum secretion. The problem with this approach is the pulsatile nature of progesterone secretion and the tremendous variance one encounters when extrapolating from only a handful of values.
|
For these reasons, any comparisons of serum hormone and TEB methods without the more acceptable standard of total integrated progesterone in the luteal phase would be equivocal. Alternative diagnostic methods, including sonography, BBT charting, and urine or saliva sampling, have received only mixed reviews. The advantages and disadvantages of each method are discussed.
As previously noted, the lack of a gold standard for the diagnosis of LPD has always plagued clinicians (Fig. 16). Logically, if one could measure serum levels of progesterone at frequent intervals throughout the luteal phase, one could best assess corpus luteum function. What sampling interval is sufficient to overcome the effects of daily fluctuation, and what are the effects of other secretory products of the corpus luteum? Even the measurement of progesterone on a daily basis, as suggested by Jones, is a compromise of sorts. The ultimate test of luteal sufficiency would be to perform frequent sampling in a conceptive cycle. Alteration of the corpus luteum by trophoblastic hCG early in gestation affects progesterone secretion, however, precluding any meaningful assessment. As a result, most investigations that appear in the medical literature compare one diagnostic test with another equally unvalidated method. This circular reference pattern brings into question the true incidence of the disorder and the sensitivity and precision of available tests. The inherent limitations of each of the tests available for LPD should be understood before using them for clinical decision making.
|
Endometrial Biopsy
Despite claims that the endometrial biopsy is the gold standard against which other diagnostic means should be measured,135 at best it should be regarded as a “bronze” standard. No gold standard exists, and the biopsy method is fraught with too many inconsistencies to be considered a “silver” standard. The criteria for endometrial dating first appeared in the classic article by Noyes and colleagues136 in the first issue of Fertility and Sterility in 1950. Its importance as a landmark report in the diagnosis and treatment of infertility is exemplified by its status as the most frequently cited article in the history of that journal.137 A total of 8000 biopsy specimens were taken to establish norms for each day in an idealized 28-day menstrual cycle. Although the massive sample size involved in this work and its later follow-up report138 are indisputably valuable, some flaws have been recognized, including (1) the biopsy specimens came from a skewed population with infertility, (2) validation was by BBT charting, and (3) interobserver variation for histologic dating was within 1 day only 63% of the time and within 2 days 81% of the time.81
If one chooses to perform a TEB for luteal assessment, it is crucial to understand the controversial questions that critics and proponents alike have posed regarding this technique. The first question concerns the optimal time in a menstrual cycle to perform the biopsy. Because the crucial window of implantation is between postovulatory days 6 and 10, logic dictates that sampling of the endometrium during this time would have the greatest predictive value. Past investigators argued, however, that early and mid luteal biopsy specimens result in wider variation in histologic dating than do late luteal biopsy specimens when dated according to the next menstrual period.6,139,140,141 This dating technique, first touted by Noyes and colleagues in their original article,136 involves numbering days backward, assuming an idealized 28-day cycle, regardless of the true cycle length. Others have endorsed late luteal TEB because it should reflect more accurately the response of the endometrium to the total hormonal output of the corpus luteum after ovulation.64,142 Other investigators have shown no increased variation in dating when biopsy specimens were obtained in the mid luteal phase and dated using mid cycle events as the reference point.143,144,145,146 This latter dating method involves identification of the urine or serum LH surge or alternatively sonographic follicle collapse to number the luteal days subsequently.
Further support for mid luteal biopsy came from Castelbaum and colleagues,147 who performed two luteal phase biopsies in the same cycle in 33 infertile women. Using the mid cycle urinary LH surge as a reference point, they detected an out-of-phase endometrium twice as often in the midluteal biopsy specimen as in the late luteal biopsy specimen. Depending on the pathologist, 80% to 100% of the abnormal mid luteal endometria had corrected spontaneously by the time the late luteal phase occurred. A higher detection rate does not equate to a higher rate of disease, given the general lack of validation for TEB in any part of the luteal phase. This striking finding does serve to heighten, however, the skepticism of past results from LPD studies that relied on late luteal biopsy specimens.
The second crucial point for clinical interpretation of TEB results is the method selected to judge normalcy. Some investigators termed out-of-phase to mean 3 or more days different from the reference date; others have used 2 or more days as the criterion. The importance of this point was illustrated by Davis and associates9 in a study of five regularly menstruating, fertile women who underwent a total of 39 endometrial biopsies over a 1-year period. Changing the criterion from 2 or more days to 3 or more days to be considered out-of-phase altered the incidence of sporadically abnormal biopsy specimens from 51.4% to 31.4%. Similarly the incidence of sequential pairs of out-of-phase biopsy specimens decreased from 26.7% to 6.7%. The incidence figures they reported using the more restrictive criterion are similar to those of the infertile population at their center and the 5% to 10% incidence figure for LPD as reported by others.
A third consideration is how many biopsy specimens should be taken before a clinician can make the diagnosis of LPD confidently? In 1980, Wentz6 reported a study of biopsies performed in two separate menstrual cycles that showed 19% of women with an initial abnormal biopsy specimen had a second normal biopsy specimen; 33% of women with a normal first biopsy specimen had a second abnormal biopsy specimen. Balasch and coworkers148 performed a study with three luteal biopsy specimens in 60 infertile women over a 1-year period. Although the biopsies were not performed in sequential cycles, they showed that 16% of women had only one abnormal biopsy specimen, 15% had two abnormal biopsy specimens, and 27% had three abnormal biopsy specimens. In an attempt to avoid a false-positive result, the current practice of obtaining two consecutive out-of-phase biopsy specimens before making the diagnosis of LPD has evolved. This approach has not been tested rigorously and is considered to be a compromise.
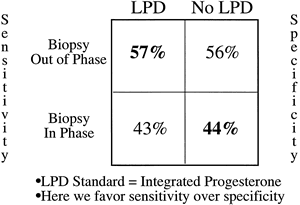
The last question for meaningful interpretation of TEB is how does this method compare with established standards? If one considers the silver standard to be integrated serum progesterone during the luteal phase, only a handful of comparisons are available in the literature. Batista and colleagues149 published a report of 50 normal women who underwent daily blood sampling in a menstrual cycle in which a mid luteal biopsy specimen was obtained. Using a criterion of 3 or more days of delay as abnormal, they found a significant correlation with integrated luteal progesterone. They also showed that only 2 of 14 abnormal biopsy specimens were accompanied by low integrated progesterone levels. In a study by Jordan and associates,150 integrated progesterone was compared with late luteal biopsy specimen using 3 or more days as the cutoff for delay (Fig. 17). When the mid cycle reference method of endometrial dating was used, sensitivity and specificity for biopsy were 29% and 56%. When dating from the next menstrual period, these variables changed to 57% and 44%. Both of these studies were hampered by relatively few subjects with low integrated progesterone levels, but they illustrate the considerable overlap that one sees in progesterone output for women with and without out-of-phase biopsy specimens.
|
Intuitively the ultimate or “platinum” standard should be the performance of endometrial biopsy in predicting the outcome of conceptive cycles. Multiple studies in which TEB was performed in early gestation during the presumed late luteal period consistently show that histologic delay of the endometrium is associated with normal rates of viable pregnancy.151,152,153,154,155 Because there was no ensuing menses in these cycles, dating was performed using mid cycle events, such as BBT. One report found higher rates of in-phase TEB when performed before luteal day 12 (i.e., mid luteal).151 Biopsy specimens taken after luteal day 12 showed persistent stromal edema typical of early gestation, which may account for the perceived histologic delay in earlier reports. In general, the rates of spontaneous abortion in biopsied cycles were not increased over those for the general infertility population. For these reasons, luteal biopsy during conceptive cycles is of little predictive value and should be avoided.
Future uses of endometrial biopsy specimens may include identification of integrins and other cell surface glycoproteins involved in attachment of the blastocyst. A well-timed mid luteal biopsy specimen with evidence of the ανβ3 integrin may indicate adequate uterine receptivity, obviating the need for serum measurements or subjective histologic dating. The reliability and cost-effectiveness of such measures have not yet been established.
Serum Progesterone
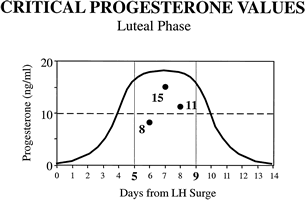
Because of the pulsatile nature of progesterone secretion, single serum measurements during the luteal phase are of little or no value in diagnosing LPD.2,156 Numerous studies have sought to prove or disprove the sensitivity of single measurements for the diagnosis of LPD, usually in conjunction with endometrial biopsy as the standard. The results have been too erratic to allow meaningful interpretation. Some investigators have attempted to extrapolate from single values to calculate integrated luteal progesterone, but these results are subject to the same variation and should be viewed with skepticism.157 Several investigators have tried to pool multiple samples to find an estimate closer to the true mean value. Abraham and colleagues158 suggested the value of taking three samples from the mid luteal phase for accurate assessment of corpus luteum function. In a more elaborate study that compared multiple diagnostic means with integrated serum progesterone levels, Jordan and associates150 showed that the sum of three random progesterone measurements made between luteal days 5 and 9 had a sensitivity of 100% and specificity of 80% when 30 ng/mL was used as a cutoff (Fig. 18). It seems important that these samples be taken in the mid luteal phase because results using a wider interval of days (luteal days 4 to 11) were not as favorable. This also may explain in part why other investigations using multiple samples have reported equivocal results. The other reason for discrepant findings probably stems from the use of TEB as the standard in these studies.
|

Other Methods
Various other modalities have been investigated to aid in the diagnosis of LPD. Studies of normal17 and infertile159 populations have suggested that about 5% of reproductive-age women have short luteal phases (≤11 days), but no significant increase among infertile women was found compared with normal women. Others have used BBT charting to assess the luteal phase in cycles with histologically proven LPD. Despite considerable overlap with the normal biopsy group, women with out-of-phase biopsy specimens do tend to have shorter luteal phases, although no association was found for the rate of temperature rise and LPD.160 When used in cycles with LPD as diagnosed by low integrated progesterone, the BBT had a poor sensitivity of 14% (Fig. 19).150 It is likely that women who experience a luteal phase of short duration have corpus luteum dysfunction, but this probably represents a more severe form of LPD that occurs in a few patients with the disorder. The BBT may be helpful as a screening tool in this case but should not be relied on as the sole diagnostic method for LPD.
|

Ultrasound has been evaluated as a means of detection of abnormal folliculogenesis preceding inadequate corpus luteum formation. Geisthovel and coworkers161 reported a smaller mean preovulatory follicle in women with LPD (BBT as the standard) compared with normal cycles (17.7 ± 2.9 mm versus 23 ± 2.3 mm). Check and associates60 studied 50 women with biopsy-proven LPD and found that 40% of them ovulated from follicles of less than 17 mm. They also found an increased association between LPD and the luteinized unruptured follicle syndrome, in which serum endocrine profiles change normally in the luteal phase despite a persistently enlarging ovarian follicle. Subsequent studies using histologic and integrated serum progesterone criteria for the diagnosis of LPD failed to show any significant difference in follicle size compared with normal cycles.49,150 Others have looked to prove differences in ovarian blood flow in LPD cycles using color and pulsed Doppler techniques.162,163 Results are conflicting but show an overall trend toward diminished intraovarian blood flow in LPD cycles. Until problems with interobserver variation and needs for technical expertise are addressed, sonography is not recommended as a standard diagnostic test for LPD.
Other alternative diagnostic measures that have been investigated to avoid the invasiveness of biopsy or the variability in serum sampling include measurement of urinary pregnanediol, salivary progesterone levels, and serum progesterone-associated endometrial protein. Urinary pregnanediol is a metabolite of progesterone that presumably is not subject to as much variability as serum levels. Although pregnanediol concentrations are measured in single specimens, they represent the sum total of progesterone breakdown products since the time of the last void (i.e., physiologic pooling). Several studies comparing its efficacy with endometrial biopsy164 and integrated serum P165 have shown promise, but large-scale clinical trials are lacking. Collection of daily saliva samples during the luteal phase for integrated progesterone is an excellent way to avoid the pitfalls of single sampling while avoiding an invasive procedure such as biopsy. Reports of concomitant use of serum and saliva samples show excellent correlation despite wide variation.166,167 Integrated salivary progesterone levels have no proven predictive value, however, in women with histologic LPD and a history of recurrent miscarriage.168 Likewise, a large clinical trial using integrated serum progesterone as the standard for LPD showed poor predictive value for single or multiple mid luteal salivary progesterone measurements.165 Progesterone-associated endometrial protein is secreted by the endometrium and has been associated with histologically diagnosed LPD, but validation in larger clinical trials is lacking.169
Diagnostic Recommendations
In the evaluation of possible LPD in the clinical setting, it is important to keep in mind five factors: patient selection, test sensitivity and specificity, invasiveness, convenience, and cost. In general, routine LPD screening of all patients presenting with infertility is unnecessary. Greater predictive value for testing arises when the group studied has a higher prevalence of disease. In a general infertility population, testing is best limited to patients with otherwise unexplained infertility, advanced maternal age, recurrent abortion, strenuous exercisers, underweight women or women with recent weight loss, women with occasional amenorrhea, and women with galactorrhea. Patients who have documented short luteal intervals by either BBT or detection of a urinary LH surge also should be tested. Further consideration of diagnostic testing for LPD should be given to patients who present to a referral infertility practice who have failed prior empirical treatment modalities.
Historically the dogma regarding the diagnosis of LPD always has been to assess luteal function in a minimum of two cycles before making the diagnosis. In the group of women who are at high risk for LPD, however, it would be reasonable to begin treatment after one representative cycle after discussing the risks and benefits of treatment with the patient (see later). All women diagnosed with LPD should undergo further evaluation with serum thyroid-stimulating hormone and prolactin because thyroid disease and hyperprolactinemia are common disorders that are amenable to specific treatment, and they can affect luteal function in women of reproductive age.
For reasons noted earlier, clinicians in our medical center choose to test for LPD by measuring the concentration of progesterone in the pooled serum from three blood samples drawn between luteal days 5 and 9 (Fig. 20). Using data from clinical trials in our laboratory,150 we determined that a pooled concentration of 10 ng/mL or more is sufficient to rule out LPD. Patients with a persistent value (two or more cycles) less than 10 ng/mL should be treated. This method seems to be the most sensitive, while eliminating the sometimes equivocal interpretation associated with TEB. Although this method requires phlebotomy on 3 separate days, a moderate amount of patient motivation overcomes this small hurdle. Patients consider this method to be physically less invasive than TEB, even though the latter offers the convenience of fewer clinic visits. Total laboratory costs and patient fees also tend to be less using pooled luteal progesterone levels. In many centers, analysis of three separate serum samples without pooling is still more cost-effective than TEB. In this situation, the sum of three serum progesterone samples should exceed 30 ng/mL.
|
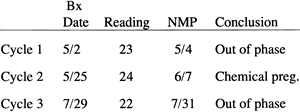
In treatment cycles, whether they include ovulation induction or progesterone supplementation, the best diagnostic test for LPD is TEB. Despite the aforementioned shortcomings of TEB, most treatment modalities create supraphysiologic levels of serum progesterone, for which there are no established normal values. Serum progesterone levels vary with the dosing schedule when using progesterone supplementation for treatment. For this reason, TEB should be used as a next-best alternative to verify the adequacy of treatment. Most investigations using TEB have used a lag of 3 days or more to define out-of-phase, with dating based on the first day of the next menstrual period as day 28, by convention. For example, a patient has a TEB on January 1 that subsequently was read as histologic day 23. Her next menstrual period began on January 3, which would be considered day 28. Counting backward, her January 1 biopsy specimen should have been consistent with day 26 findings, but it was 3 days out-of-phase and abnormal. This patient would be considered to have a histologic delay of her endometrium indicative of LPD (Fig. 21). This dating method is the one most commonly used, although it is acceptable to date biopsy specimens according to mid cycle events.170
|
TREATMENT
The treatment of LPD has fostered nearly as much controversy as the incidence and diagnosis have. Much of this controversy has to do with the dearth of prospective randomized trials showing the efficacy of any treatment modality for this disorder. In an extensive review of the literature, Karamardian and Grimes171 exposed the flaws of nearly all of the articles published before 1992 regarding the treatment of LPD and concluded that insufficient evidence exists for selection of a particular treatment. Balasch and colleagues172 published the only prospective randomized trial comparing two forms of treatment, progesterone vaginal suppositories and oral dehydrogesterone, with no treatment for women with LPD as diagnosed by TEB. They defined success as a return to normal histology or term pregnancy. Using these criteria, women treated with progesterone suppositories and dehydrogesterone had a successful response in 62.5% and 68.7% of cases (not significant). These figures were significantly different from the group without treatment, who experienced a 16.6% success rate (p < .001 by chi-square test). This study has been criticized for its lack of statistical power, which was due in great part to its small sample size (44 women). Also, no statistical difference was found between groups when pregnancy rate alone was used as the sole criterion for success. Given the ethical dilemma of withholding treatment for LPD for the sake of a placebo-controlled study design when a perceived benefit exists, it is unlikely that further proper scientific validation will ever exist.
It is pertinent to review the potential benefits of the various forms of therapy with an understanding of the limited claims that can be made regarding efficacy. Several observational reports of pregnancy rates before and after treatment for LPD warrant discussion, however.
Ovulation Induction
Ovulation induction has long been a part of the therapeutic armamentarium for LPD. Use of CC, gonadotropins, or both to stimulate multifollicular growth is known to increase steroid hormone levels (estradiol and progesterone) throughout the menstrual cycle, which usually stimulates adequate folliculogenesis and induction of steroid receptors. Ovulation induction also may serve to offset the imbalance of LH and FSH that has been implicated as a causative factor in some cases of LPD. It is paradoxical that gonadotropins and CC also have been implicated as causative agents for LPD. This implication may be rationalized by appealing to the multifactorial cause of the disorder. For women who develop corpus luteum insufficiency as a result of inadequate folliculogenesis, controlled ovarian stimulation may overcome subtle defects in follicle formation. If patients have an abnormality of small or large luteal cell function, they may not respond to enhancement of follicular-phase gonadotropin levels.
In a retrospective study on the use of gonadotropin stimulation for the treatment of biopsy-proven LPD in women with recurrent miscarriage, Li and colleagues173 reported significantly lower miscarriage rates for treatment cycles versus control cycles (2 of 11 [15%] versus 7 of 12 [58%]; p < .05). They also found reversion to normal histology in 85% of women treated with gonadotropins during cycles in which subjects used barrier contraception. Although small, this investigation highlights the ability of follicular-phase intervention to improve the luteal phase environment, although the exact therapeutic mechanism is unclear.
In general, because exogenous gonadotropins offer more potent stimuli to the ovaries than CC, they are more likely to overcompensate for the underlying defect. For this reason and cost and risk issues, CC is the first-line ovulation induction treatment, using the lowest effective dose that normalizes luteal parameters. A typical CC regimen would be 50 mg daily for 5 days beginning on cycle days 3 to 5. In LPD cases in which mild hypothalamic dysfunction is suspected, such as in women who are less than their ideal body weight, doses of 25 mg daily taken for 5 to 10 days may be more efficacious, avoiding undesirable effects of CC therapy, such as diminished cervical mucus quality. Because normal values for luteal progesterone in CC cycles have never been unequivocally established, TEB is the mainstay for evaluation of therapy.
Exogenous Progesterone Supplementation
Exogenous progesterone supplementation of the luteal phase is the most common treatment of LPD. Although early reports of the use of synthetic progestins showed no associated teratogenicity with first-trimester exposure,174 supplementation has been limited almost exclusively to natural progesterone preparations, not synthetic progestins. This is due in part to evidence that the latter suppress endogenous progesterone secretion after ovulation.175 Extensive retrospective reviews of experience using natural progesterone preparations have failed to show any increase in congenital anomalies over baseline incidences.176,177 Early reports of congenital anomalies secondary to progestins have been disproved by subsequent, more extensive studies.178
Results of hormone replacement cycles in women scheduled to receive embryos from donor oocytes provide excellent insight into the absolute needs for proper nidation. These women are typically menopausal or in a state of pseudomenopause brought about by the use of GnRH analogues, such as leuprolide acetate. They offer a “clean slate” on which various combinations of medications may be “stacked” to create an optimal fertile environment. What is immediately clear from such experiments is that replacement of progesterone after first priming the endometrium with estrogen is enough to allow and sustain pregnancy, despite omission of other luteal phase products. A summary of such studies (Table 2) shows the superiority of intramuscular progesterone and vaginal preparations as opposed to oral micronized progesterone.187 Although estrogen replacement in such cycles is typically within the physiologic range, direct inferences made from these cycles for LPD cycles is difficult secondary to underlying endogenous progesterone production in the latter. Still, an understanding of the pros and cons of each form of therapy may influence decisions depending on the clinical scenario.
TABLE 2. Summary of Studies Comparing Hormonal and Histologic Parameters Between Different Routes of Progesterone Administration as Luteal Phase Support in Artificial Cycles for Oocyte Donation After Pretreatment with Various Types of Estrogen
| Author | Cause | N | Daily P Dose | Luteal P Values | Day of Biopsy | Biopsy Results |
| Dehou (1987)179 | Primary ovarian failure | 8 | MP 300–600 mg p.o. | 16–24 | Maturation delay | |
| 10 | P in oil 50–100 mg i.m. | 16–24 | Normal glandular maturation. Fibrocystic stroma in first cycle, improved in next cycles | |||
| Devroey (1989)180 | Absent ovaries | 11 | MP 300 mg p.o. (100 mg on day 14) | Lowest serum P between all four groups | 21 | 100% out of phase |
| 31 | P in oil 100 mg i.m. (50 mg on day 14) | Serum P five times higher than the vaginal groups | 21 | 2/31 out of phase | ||
| 18 | MP 300 mg p.v. (100 mg on day 14) | 21 | 18/18 in phase | |||
| 10 | MP 600 mg p.v. (200 mg on day 14) | 21 | 10/10 in phase | |||
| Critchley (1990)181 | Premature ovarian failure | 5 | MP 300 mg p.o. | 14 ± 2 (mean ± SE) nmol/L* | 21 | 1/5 normal |
| 4 | 100 mg MP (p.v. pessary) | 30 ± 16 (mean ± SE) nmol/L* | 21 | 2/4 normal | ||
| 6 | 300 mg MP p.v. | 45 ± 5 (mean ± SE) nmol/L* | 21 | 5/6 normal | ||
| Bourgain (1990)182 | Primary ovarian failure | 12 | MP 300 mg p.o. | 2.79 ± 0.6 (mean ± SEM) ng/mL | 21 | 1/12 in phase |
| 34 | P in oil 100 mg i.m. | 43.4 ± 0.60 (mean ± SEM) ng/mL† | 21 | 16/34 in phase | ||
| 21 | MP 300 mg p.v. | 6.79 ± 1.28 (mean ± SEM) ng/mL† | 21 | 16/21 in phase | ||
| 8 | MP 600 mg p.v. | 8.09 ± 1.74 (mean ± SEM) ng/mL† | 21 | 6/8 in phase | ||
| Davies (1990)183 | Premature ovarian failure/gonadal dysgenesis/ | 22 | P in oil 25–50 mg i.m. | 60 ± 8 nmol/L† | 21 | Mean endometrial score: −1.5 days§ |
| 11 | P pessary 200–400 mg p.v. | 29.8 ± 5.4 nmol/L† | 21 | Mean endometrial score: −2.9 days§ | ||
| Sauer (1991)184 | Idiopathic ovarian failure/postsurgery/radiation/ | 19 | P in oil 100 mg i.m. (50 mg on day 15) | 48.8 ± 10.4 ng/mL | 26 | 19/19 normal |
| 19 | P suppositories 200 mg p.v. (100 mg on day 15) | 29.4 ± 4.8 ng/mL | 26 | 7/19 excessive estrogen effect | ||
| Miles (1994)185 | Functionally agonadal | 5 | P in oil 100 mg i.m. | 69.8 ± 5.9 (mean ± SE) ng/mL∥ | 21 | Mean dating of glands: 18.9 ± 0.1 days |
| 15 | MP 800 mg p.v. | 11.9 ± 1.2 (mean ± SE) ng/mL∥ | 21 | Mean dating of glands: 20.5 ± 1 days | ||
| Gibbons (1998)186 | Primary ovarian failure/diminished ovarian reserve | 24 | P in oil 100 mg i.m. | 89.3 ng/mL¶ | 25–27 | 23/24 in phase |
| 55 | P gel 180 mg p.v. | 19 ng/mL¶ | 25–27 | 55/55 in phase |
P, progesterone; MP, micronized progesterone; p.o., orally; p.v., vaginally; i.m., intramuscularly.
*P < .01.
†P < .0001.
†P = .014.
§P < .05.
∥P < .05.
¶P < .00001.
Adapted from Tavaniotou A, Smitz J, Bourgain C, Devroey P: Comparison between different routes of progesterone administration as luteal phase support in infertility treatments. Hum Reprod Update 6:139, 2000.
Oral micronized progesterone offers the most convenient form of luteal supplementation; however, its use may be associated with systemic side effects, such as sedation, flushing, and nausea. After ingestion, progesterone is absorbed rapidly and metabolized by the intestines, later reaching the liver where glucuronyl and sulfuryl transferases render it hydrophilic and more easily excreted. A single 100-mg dose can be expected to cause a Cmax in serum of 6.5 ng/mL within 2 to 4 hours in menopausal women, with higher doses leading to proportionate serum increases.188,189 These levels are highly influenced by food intake and may be more than fivefold higher when taken immediately after a meal.188 In addition, serum levels of progesterone remain significantly elevated for only 6 to 7 hours after an oral micronized dose, necessitating more frequent dosing to sustain levels.189,190,191 Clinicians should bear in mind that micronized progesterone is prepared in an oil base, in most cases peanut oil, which can be highly allergenic. Controversy exists regarding the true progesterone effect on the endometrium at a given serum concentration after oral micronized progesterone ingestion because some investigators have shown assay interference from high concentrations of progesterone metabolites.192 In studies of IVF cycles in which high doses of oral micronized progesterone have been given for luteal support, circulating metabolites have been blamed for decreased implantation rates compared with cycles using intramuscular progesterone with similar serum concentrations.193 Despite these shortcomings, the ability of a single daily dose of oral micronized progesterone to induce secretory change of an estrogen-primed endometrium during hormone replacement therapy studies is a testimony to its end-organ effect. It may provide the most efficacious means for “fine-tuning” serum progesterone in cases in which only modest boosts are required.
Vaginal progesterone delivery offers several advantages over oral progesterone, most notably, avoidance of first-pass metabolism in the gastrointestinal tract and liver with a lower incidence of sedation. Patients may experience side effects, such as a discharge, irritation, or local feelings of warmth, but generally find them to be acceptable.194,195,196 Serum concentrations reach a maximum at 3 to 8 hours, depending on the formulation used, with a slow decline to baseline after 8 hours.191,195,197 More importantly, vaginal administration of progesterone leads to what has been deemed a first-pass uterine effect, that is, higher endometrial tissue concentrations of progesterone than one would anticipate based on serum levels.198 Progesterone vaginal suppositories in doses of 25 to 50 mg or micronized progesterone 100- to 200-mg capsules may be inserted into the vagina twice daily beginning 3 to 4 days after presumed ovulation, timed either by BBT or by urine LH testing. The main disadvantages of suppositories are the lack of standardized commercial preparations and the undesirable discharge they can create for the patient. In one of the only prospective, randomized studies comparing oral and vaginal progesterone in non-IVF, CC ovulation induction cycles, vaginal progesterone cycles were marked by lower pregnancy rates and higher insulin-like growth factor binding protein-1 (IGFBP-1) serum concentrations.199 Although subjects were anovulatory and did not have a diagnosis of LPD before treatment, the relative endometrial potency of the two forms of progesterone delivery may account for the differences and may not reflect the true effect in LPD patients.
As an alternative, a newer intravaginal formulation of natural progesterone in a water-soluble, polycarbophil gel that adheres to the vaginal mucosa may be used, causing considerably less discharge with equal or better tissue levels in the endometrium.198 In doses of 90 mg/day, the sustained release of progesterone afforded by this vehicle offers excellent results in donor oocyte IVF cycles.200 As is true for all forms of vaginal progesterone delivery, measurement of circulating hormone levels is not useful in monitoring treatment efficacy. The exact dose of progesterone gel for the most effective treatment of LPD has yet to be determined; however, commercially available preparations of 45 and 90 mg are available in North America.
Another method of supplementation is daily intramuscular injection of 12.5 to 25 mg of progesterone in oil. Although this method is the most uncomfortable option for patients, a single 25-mg injection provides luteal phase serum levels of progesterone, with measurable elevations 36 hours later.197 Its clinical efficacy as luteal support in IVF cycles is unmatched; however, injection-site pain and localized inflammation and induration have limited the widespread use of progesterone in oil as a supplement for LPD cycles.
Other Forms of Luteal Support
In addition to the use of progesterone for supplementation, some clinicians have chosen to administer hCG intramuscularly. Exogenous hCG, because of its structural homology with LH, stimulates the corpus luteum to increase steroid hormone production. Various doses and dosing intervals have been used in assisted reproductive cycles, most commonly 2500 to 5000 U every 3 days after ovulation. Its use in LPD cycles is limited. The disadvantages of this technique include the need for repeated injections and the inability to detect early pregnancy reliably with serum hCG levels. In addition, in cases of LPD in which a small cell defect predominates, hCG may have little or no impact on luteal function. Experience with ovulation induction in which GnRH agonists were used for down-regulation has shown an increased risk of ovarian hyperstimulation syndrome with hCG luteal support.114,201,202 For these reasons, hCG is no longer the treatment of choice for luteal support of either spontaneous or stimulated cycles.
Therapies for LPD specifically targeted to the central nervous system have shown promise in the research setting. The ability to induce normal ovulatory menstrual cycles in women with hypothalamic amenorrhea using a GnRH pump led investigators to apply this treatment to women with LPD.203 Five women with LPD (TEB diagnosis) were treated with pulsatile GnRH (5 μg intravenously every 90 minutes) beginning on days 1 to 4 of the cycle, with continuation until the next menses or pregnancy. One patient conceived during 1 of the 12 treatment cycles; all others experienced improvement in endocrine or histologic parameters. The use of this therapy clinically is limited because of cost, invasiveness, and the capability of achieving comparable pregnancy rates using CC or gonadotropins. Other investigators have shown resolution of LPD and increased fecundity with treatment with nonhormonal drugs that act on the central nervous system, such as diphenylhydantoin and nialamide.204 Although neither of these therapies has been accepted widely clinically, they illustrate the importance of normal central nervous system mechanisms in the establishment of adequate folliculogenesis and subsequent corpus luteum function.
Bromocriptine is a dopamine agonist with central nervous system activity used to inhibit secretion of prolactin from the anterior pituitary. In theory, it may be useful in lowering the subtle elevations in prolactin that are difficult to detect with random serum measurements and that may contribute to the development of LPD. In practice, bromocriptine should be prescribed (1.25 mg twice daily) only to women with documented hyperprolactinemia to avoid the undesirable risks and side effects of the medication.
Results of Treatment
Several uncontrolled studies that report pregnancy rates after treatment of LPD describe acceptable fecundity rates compared with a fertile population and offer compelling evidence for therapy. The first report by Daly and colleagues205 involved 33 infertile women with LPD diagnosed by two abnormal TEBs (Fig. 22). Their patients initially were treated with progesterone vaginal suppositories (25 mg twice daily) with repeat TEB performed in the first treatment cycle to judge efficacy. Women continued on this regimen for six cycles regardless of TEB findings. Women who failed to conceive were switched to CC (50 mg daily) on cycle days 5 to 9, again with TEB, and this regimen was continued for 6 months regardless of TEB findings. Women who failed were treated with CC with progesterone, gonadotropins, or dexamethasone if appropriate. Following this treatment algorithm, 81% of compliant patients conceived and had viable pregnancies. Using life-tablele analysis, they showed normalization of cycle fecundity and cumulative pregnancy rates when TEB was corrected. Although fecundity for patients treated with progesterone was higher than for those treated with CC, a significant negative selection bias against CC existed because only patients who failed progesterone treatment were included in this treatment group.
|

Solid evidence for the effect of treatment for LPD also came from a study by Murray and coworkers,206 who used vaginal progesterone or CC for their patients. Patients were continued on one type of therapy if TEB was corrected. Overall mean follow-up time for both groups was 6 months. Using life-tablele analysis, they showed a cumulative pregnancy rate at 6 months of 58% for CC-treated patients and 49% for progesterone-treated patients compared with an estimated normal rate of 74% (Fig. 23). By 12 months of follow-up, 81% of CC-treated patients and 100% of progesterone-treated patients were pregnant compared with an estimated normal rate of 93%. No significant difference was detected between the two treatment groups. In both studies, cumulative pregnancy rates for patients with corrected LPD were similar to rates projected for a normal, fertile population, supporting treatment of LPD.
|

For the subgroup of LPD patients who suffer from recurrent miscarriage, several reports present successful series of patients treated with progesterone or CC. A retrospective review by Tho and associates207 of 100 couples who presented with recurrent miscarriage at their institution identified 23 couples for whom abnormal TEB (LPD) was the sole abnormality. Twenty-two couples were treated with luteal phase progesterone supplementation; the remaining couples were treated with CC and progesterone. Of the 23 treated patients, 21 (91%) subsequently carried their pregnancies to term. An extensive meta-analysis of controlled trials of progesterone support for pregnancy in women with recurrent miscarriage by Daya208 identified three studies in the literature that were random or quasi-random with placebo controls. Although none of these studies specifically diagnosed LPD before instituting progesterone therapy, the recognized incidence of LPD in this group (32.5% to 60%) allows reasonable inferences to be made. By combining these three studies, Daya208 found an overall odds ratio for treatment of 3.09 (95% confidence interval, 1.28 to 7.42). Taken together, the prevailing evidence suggests that treatment can correct inadequate corpus luteum function despite the lack of well-designed studies.
TREATMENT RECOMMENDATIONS
Selection of Treatment
At present, selection of a particular treatment modality in our center depends on the presumed cause of LPD. Most obvious are women with hyperprolactinemia or abnormal thyroid function; they should be treated appropriately to correct these disorders, which also adequately treats LPD in most instances. CC rarely is used as a first-line treatment because of its potential to induce LPD, but it is the treatment of choice for women with abnormally short or long follicular phase length and for women who have sonographic documentation of ovulation from follicles of less than 20 mm mean diameter. In these instances, the cause of LPD seems to be inadequate folliculogenesis, which is generally more refractory to progesterone supplementation. In most of our patients, administration of natural progesterone in the luteal phase has been the first line of therapy. We use oral micronized progesterone, 100 mg three times daily, beginning 4 days after the detection of a preovulatory urine LH surge or an ovulation-triggering injection of hCG. For patients who experience side effects related to oral progesterone, we administer 200 mg of micronized progesterone vaginally twice daily. The less frequent dosing interval is possible because of prolonged tissue levels using this route of delivery. Daily application of 4% vaginal progesterone gel (45 mg) holds promise despite the lack of clinical data but costs considerably more than either of the aforementioned options.
Onset and Duration of Treatment
Because the survival of the blastocyst depends on adequate implantation in the secretory endometrium, intuitively the time to begin treatment is early in the luteal phase (several days after ovulation). Withholding treatment until documentation of pregnancy (2 to 3 weeks after ovulation) does not have the same beneficial effects on nidation, although adequate trials comparing early with late onset of treatment are lacking. In our institution, we begin progesterone supplementation 3 to 4 days after the presumed day of ovulation to avoid suppressing periovulatory cervical mucus and to maximize the effect of progesterone on the endometrium before implantation. This translates into starting treatment 4 days after a positive urine test for LH or an artificial surge using exogenous administration of hCG (5000 to 10,000 U) during monitored cycles. If no pregnancy results in a particular treatment cycle, progesterone supplementation is discontinued with the onset of menses, with reinstitution in the luteal phase of the next cycle. The duration of treatment should be at least three but not more than six cycles without a re-evaluation of the patient.
If pregnancy is confirmed with a positive test for β-hCG in the serum or urine, treatment is continued through the 10th week of pregnancy. This recommendation is based on studies of luteectomy in early pregnancy in women scheduled to undergo therapeutic abortion.209,210 These studies demonstrated that luteectomy at 7 weeks’ gestational age led to abortion in all pregnancies (n = 6), but at 9 weeks, progesterone levels decreased transiently with no abortions (n = 3). Exogenous progesterone delivered after lutectomy at 7 weeks preserved pregnancies. If women taking oral micronized progesterone begin to develop worsening nausea and bloating that makes their capsules less palatable, they may convert to twice-daily vaginal capsules or suppositories as alternatives.
Monitoring Treatment Effect
Because the use of CC, gonadotropins, or exogenous progesterone alters the pharmacodynamics of circulating progesterone, assessment of treatment should be performed using an endometrial biopsy. After treatment, if the biopsy specimen remains out-of-phase, an alternative treatment or, in the case of CC, a higher dose should be used. With the potential for induced LPD with CC or gonadotropins, concomitant use of progesterone may be necessary.
Pretreatment Considerations
The decision to treat or not to treat a patient is at the core of what it means to be a physician. When performing diagnostic tests and developing treatments, one always must keep in mind the principles of beneficence and nonmalfeasance—that is, whether treatment helps or harms the patient. This issue is best broached by using the technique of receiver operator characteristic (ROC) curves. This method involves plotting a test’s true positive rate (sensitivity) on the y-axis versus the false-positive rate (1 − specificity) on the x-axis using multiple, arbitrary threshold values to define abnormality (Fig. 24). The resulting curve bends toward the upper left corner of the graph, with an initial slope near infinity close to the y-axis and a later slope close to 0 away from the y-axis. The optimal cutoff point for a positive test is the point on the ROC curve where the slope of ROC curve equals:
Slope of ROC curve = (net cost/net benefit) × (1 − p/p)
where the net cost is the overall cost to a patient without disease being erroneously treated and the net benefit is the potential gain to someone with disease being treated compared with remaining untreated. The other factor is the pretest probability (p), the chance that the patient has the disease before any testing begins.
The optimal cutoff point for normalcy varies from patient to patient based on these parameters (see Fig. 24). With a disease such as cancer, in which the potential net benefit of treatment is great but the cost of erroneously treating is as great or greater, one would like to have a cutoff point on the curve where the slope is steep (i.e., few false-positive results). The opposite is true in the case of LPD: The potential benefit of treatment with progesterone (i.e., pregnancy) is tremendous, but the concomitant risk of overtreatment (i.e., nausea, somnolence) is relatively low. If we arbitrarily assign a net cost of 1 and a net benefit of 10 to LPD and assume a prevalence of disease in an infertile population of about 10%, our optimal cutoff point for normalcy would be at the point on the curve where the slope is 0.9 (i.e., on the far right of the curve). Our threshold value for making the diagnosis of LPD should be set to allow a relatively high percentage of false-positive results based on the estimates used in this scenario. For some patients and some forms of treatment, these ratios and pretest probabilities are not applicable. Several excellent reviews on the ROC concept are available.211,212,213 The underlying message is that suspicion of LPD (i.e., high pretest probability) and borderline diagnostic testing should be enough compelling evidence for treatment. Although some would argue against testing in favor of empirical therapy, the latter is discouraged because no treatment is without some detrimental effect.
|
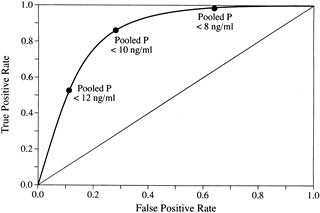
SUMMARY
LPD is a subtle disorder of corpus luteum function and has a multifactorial cause (Fig. 25). Current evidence in the medical literature confirms its existence, but investigations to date have failed to characterize its true incidence conclusively. The diagnosis is best made by measuring serum progesterone levels daily throughout the luteal phase. This method is impractical, however, except in a research setting. The best means of estimating luteal phase function available to clinicians is three pooled mid luteal progesterone levels. Although TEB has enjoyed widespread use in past investigations of LPD, it is a suboptimal diagnostic standard that still plays an important role as a test of adequacy during treatment cycles. Despite the lack of randomized, controlled trials proving efficacy of treatment, compelling reports point to improved pregnancy rates when treatment results in normalization of TEB. In consideration of the cost-benefit equation as applied to LPD, clinicians are urged to seek out this disorder and appropriately treat it when it is present.
|
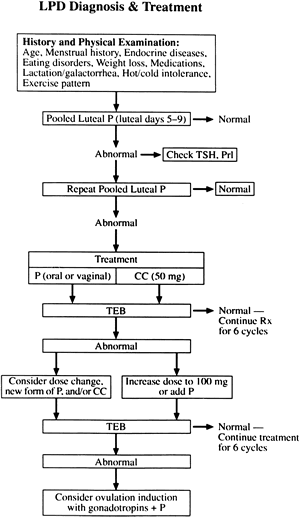
REFERENCES
Jones GES: Some newer aspects of management of infertility. JAMA 141:1123, 1949 |
|
Hayashi M, Suginami H, Taii S, Mori T: Endocrine pathophysiology of luteal phase deficiency as assessed by GnRH/TRH stimulation tests performed in the early follicular and midluteal phases of the menstrual cycle. Endocr J 40:297, 1993 |
|
Jones GS: The luteal phase defect. Fertil Steril 27:351, 1976 |
|
Balasch J, Vanrell JA: Luteal phase deficiency: An inadequate endometrial response to normal hormone stimulation. Int J Fertil 31:368, 1986 |
|
Downs KA, Gibson M: Clomiphene citrate therapy for luteal phase defect. Fertil Steril 39:34, 1983 |
|
Wentz AC: Endometrial biopsy in the evaluation of infertility. Fertil Steril 33:121, 1980 |
|
Li T, Dockery P, Cooke I: Endometrial development in the luteal phase of women with various types of infertility: Comparison with women of normal fertility. Hum Reprod 6:325, 1991 |
|
Hague WE, Maier DB, Schmidt CL, Randolph IF: An evaluation of late luteal phase endometrium in women requesting reversal of tubal ligation. Obstet Gynecol 69:926, 1987 |
|
Davis O, Berkeley A, Naus G, et al: The incidence of luteal phase defect in normal, fertile women, determined by serial endometrial biopsies. Fertil Steril 51:582, 1989 |
|
Balasch J, Creus M, Marquez M, et al: The significance of luteal phase deficiency on fertility: A diagnostic and therapeutic approach [published erratum appears in Hum Reprod 1:496, 1986]. Hum Reprod 1:145, 1986 |
|
Grunfeld L, Sandler B, Fax J, et al: Luteal phase deficiency after completely normal follicular and periovulatory phases. Fertil Steril 52:919, 1989 |
|
Peters A, Riley P, Coulam C: Prevalence of out-of-phase endometrial biopsy specimens. Am J Obstet Gynecol 166:1738, 1992 |
|
Pirke KM, Schweiger U, Strowitzki T, et al: Dieting causes menstrual irregularities in normal weight young women through impairment of episodic luteinizing hormone secretion. Fertil Steril 51:263, 1989 |
|
Gray RH, Campbell OM, Zacur HA, et al: Post partum return of ovarian activity in nonbreastfeeding women monitored by urinary assays. J Clin Endocrinol Metabol 64:645, 1987 |
|
Diaz S, Cardenas H, Brandeis A, et al: Relative contributions of anovulation and luteal phase defect to the reduced pregnancy rate of breastfeeding women. Fertil Steril 58:498, 1992 |
|
Rosenberg SM, Johnson M, Riddick DH: Luteal phase defect as a marker of imminent ovarian failure. Obstet Gynecol 59:89, 1982 |
|
Lenton EA, Landgren BM, Sexton L: Normal variation in the length of the luteal phase of the menstrual cycle: Identification of the short luteal phase. Br J Obstet Gynaecol 91:685, 1984 |
|
Apter D, Vihko R: Early menarche, a risk factor for breast cancer, indicates early onset of ovulatory cycles. J Clin Endocrinol Metabol 57:82, 1983 |
|
Bullen BA, Skrinar GS, Beitins IZ, et al: Induction of menstrual disorders by strenuous exercise in untrained women. N Engl J Med 312:1349, 1985 |
|
Cheesman KL, Cheesman SD, Chatterton RTJ, Cohen MR: Alterations in progesterone metabolism and luteal function in infertile women with endometriosis. Fertil Steril 40:590, 1983 |
|
Kauppila A, Leinonen P, Vihko R, Ylostalo P: Metoclopramide-induced hyperprolactinemia impairs ovarian follicle maturation and corpus luteum function in women. J Clin Endocrinol Metabol 54:955, 1982 |
|
Jewelewicz R, Dyrenfurth I, Warren M, et al: Effect of thyrotropin-releasing hormone upon the menstrual cycle in women. J Clin Endocrinol Metabol 39:387, 1974 |
|
Huang KE, Bonfiglio TA, Muechler EK: Transient hyperprolactinemia in infertile women with luteal phase deficiency. Obstet Gynecol 78:651, 1991 |
|
Del Pozo E, Wyss H, Tolis G, et al: Prolactin and deficient luteal function. Obstet Gynecol 53:282, 1979 |
|
Soules MR, Luteal phase deficiency. In Pitkin RM (eds): Clinical Obstetrics and Gynecology. Vo1 34:pp 123, 126 Philadelphia, JB Lippincott, 1991 |
|
Carr BR, MacDonald PC, Simpson ER: The role of lipoproteins in the regulation of progesterone secretion by the human corpus luteum. Fertil Steri1 38:303, 1982 |
|
Fritz MA, Fitz TA: The functional microscopic anatomy of the corpus luteum: The “small cell/large cell” controversy. In Pitkin RM (ed): Clinical Obstetrics and Gynecology. Vo1 34:pp 144, 156 Philadelphia, JB Lippincott, 1991 |
|
Hinney B, Henze C, Kuhn W, Wuttke W: The corpus luteum insufficiency: A multifactorial disease. J Clin Endocrinol Metabol 81:565, 1996 |
|
O’Hara A, Mori T, Taii S, et al: Functional differentiation in steroidogenesis of two types of luteal cells isolated from mature human corpora lutea of menstrual cycle. J Clin Endocrinol Metabol 65:1192, 1987 |
|
Healy DL, Schenken RS, Lynch A, et al: Pulsatile progesterone secretion: Its relevance to clinical evaluation of corpus luteum function. Fertil Steril 41:114, 1984 |
|
Filicori M, Butler JP, Crowley WF Jr: Neuroendocrine regulation of the corpus luteum in the human: Evidence for pulsatile progesterone secretion. J Clin Invest 73:1638, 1984 |
|
Rossmanith WG, Laughlin GA, Mortola JF, et al: Pulsatile cosecretion of estradiol and progesterone by the midluteal phase corpus luteum: Temporal link to luteinizing hormone pulses. J Clin Endocrinol Metabol 70:990, 1990 |
|
Schwall RH, Sawyer HR, Niswender GD: Differential regulation by LH and prostaglandins of steroidogenesis in small and large luteal cells of the ewe. J Reprod Fertil 76:821, 1986 |
|
Maas S, Jarry H, Teichmann A, et al: Paracrine actions of oxytocin, prostaglandin F2 alpha, and estradiol within the human corpus luteum. J Clin Endocrinol Metabol 74:306, 1992 |
|
Wuttke W, Jarry H, Pitzel L, et al: Luteotrophic and luteolytic actions of ovarian peptides. Hum Reprod 8(suppl 2):141, 1993 |
|
Fritz MA, McLachlan RI, Cohen NL, et al: Onset and characteristics of the mid cycle surge in bioactive and immunoactive luteinizing periovulatory ovarian steroid hormone secretion. J Clin Endocrinol Metabol 75:489, 1992 |
|
Hoff JD, Quigley ME, Yen SSC: Hormonal dynamics at mid cycle: A reevaluation. J Clin Endocrinol Metabol 57:792, 1983 |
|
Lessey BA, Yeh I, Castelbaum AJ, et al: Endometrial progesterone receptors and markers of uterine receptivity in the window of implantation. Fertil Steri1 65:477, 1996 |
|
DiZerega GS, Hodgen GD: Luteal phase dysfunction infertility: A sequel to aberrant folliculogenesis. Fertil Steril 35:489, 1981 |
|
Stouffer RL, Hodgen GD: Induction of luteal phase defects in rhesus monkeys by follicular fluid administration at the onset of the menstrual cycle. J Clin Endocrinol Metabol 51:669, 1980 |
|
Wilks JW, Hodgen GD, Ross GT: Luteal phase defects in the rhesus monkey: The significance of serum FSH:LH ratios. J Clin Endocrinol Metabol 43:1261, 1976 |
|
Aksel S: Sporadic and recurrent luteal phase defects in cyclic women: Comparison with normal cycles. Fertil Steril 33:372, 1980 |
|
Cook CL, Rao CWV, Yussman MA: Plasma gonadotropin and sex steroid hormone levels during early, midfollicular and midluteal phases of women with luteal phase defects. Fertil Steril 40:45, 1983 |
|
Sheehan KL, Casper RF, Yen SSC: Luteal phase defects induced by an agonist of luteinizing hormone-releasing factor: A model for fertility control. Science 215:170, 1982 |
|
Sherman BM, Korenman SG: Measurement of plasma LH, FSH, estradiol and progesterone in disorders of the human menstrual cycle: The short luteal phase. J Clin Endocrinol Metabol 38:89, 1974 |
|
Strott CA, Cargille CM, Griff TR, Lipsett MB: The short luteal phase. J Clin Endocrinol 30:246, 1970 |
|
Molskness TA, Woodruff TK, Hess DL, et al: Recombinant human inhibin-A administered early in the menstrual cycle alters concurrent pituitary and follicular, plus subsequent luteal, function in rhesus monkeys. J Clin Endocrinol Metabol 81:4002, 1996 |
|
Balasch J, Creus M, Vanrell JA: Luteal function after delayed ovulation. Fertil Steril 45:342, 1986 |
|
Soules MR, McLachlan RI, Ek M, et al: Luteal phase deficiency: Characterization of reproductive hormones over the menstrual cycle. J Clin Endocrinol Metabol 69:804, 1989 |
|
Andoh K, Mizunuma H, Hasegawa Y, et al: Association of immunoreactive inhibin levels with luteal function. Horm Res 37(suppl 1):37, 1992 |
|
Soules MR, Steiner RA, Clifton DK, et al: Progesterone modulation of pulsatile luteinizing hormone secretion in normal women. J Clin Endocrinol Metabol 58:378, 1984 |
|
Soules MR, Clifton DK, Cohen NL, et al: Luteal phase deficiency: Abnormal gonadotropin and progesterone secretion patterns. J Clin Endocrinol Metabol 69:813, 1989 |
|
Soules MR, Bremner WJ, Dahl KD, et al: The induction of premature luteolysis in normal women—follicular phase luteinizing hormone secretion and corpus luteum function in the subsequent cycle. Am J Obstet Gynecol 164:989, 1991 |
|
Suh BY, Betz G: Altered luteinizing hormone pulse frequency in early follicular phase of the menstrual cycle with luteal phase defect patients in women. Fertil Steril 60:800, 1993 |
|
Soules MR, Clifton DK, Bremner WJ, Steiner RA: Corpus luteum insufficiency induced by a rapid gonadotropin-releasing hormone-induced gonadotropin secretion pattern in the follicular phase. J Clin Endocrinol Metabol 65:457, 1987 |
|
Schweiger U, Laessle RG, Tuschl RJ, et al: Decreased follicular phase gonadotropin secretion is associated with impaired estradiol and progesterone secretion during the follicular and luteal phases in normally menstruating women. J Clin Endocrinol Metabol 68:888, 1989 |
|
Jones GS, Madrigal-Castro V: Hormonal findings in association with abnormal corpus luteum function in the human: The luteal phase defect. Fertil Steril 21:1, 1970 |
|
Van de Wiele RL, Bogumil J, Dyrenfurth I, et al: Mechanisms regulating the menstrual cycle in women. Recent Progr Horm Res 26:63, 1970 |
|
Soules MR, Clifton DK, Steiner RA, et al: The corpus luteum: Determinants of progesterone secretion in the normal menstrual cycle. Obstet Gynecol 71:659, 1988 |
|
Check JH, Goldberg BB, Kurtz A, et al: Pelvic sonography to help determine the appropriate therapy for luteal phase defects. Intl J Fertil 29:156, 1984 |
|
Ying Y-K, Daly DC, Randolph JF, et al: Ultrasonographic monitoring of follicular growth for luteal phase defects. Fertil Steril 48:433, 1987 |
|
Corner GWJ: The histological dating of the human corpus luteum of menstruation. Am J Anat 98:377, 1956 |
|
Harrison RT, Weir BJ: Structure of the mammalian ovary. In Zuckerman L, Weir BJ (eds): The Ovary. I. General Aspects. New York, Academic Press, 1977 |
|
Jones GS, Maffezzoli RD, Strott CA, et al: Pathophysiology of reproductive failure after clomiphene-induced ovulation. Am J Obstet Gynecol 108:847, 1970 |
|
De Moraes-Ruehsen MD, Jones GS, Burnett LS, Baramki TA: The aluteal cycle. Am J Obstet Gynecol 103:1059, 1969 |
|
Illingworth DR, Corbin DK, Kemp ED, Keenan EI: Hormone changes during the menstrual cycles in abetalipoproteinemia: Reduced luteal phase progesterone in a patient with homozygous hypobetalipoproteinemia. Proc Natl Acad Sci U S A 79:6685, 1982 |
|
Kim H-J, Kalkhoff RK: Changes in lipoprotein composition during the menstrual cycle. Metabolism 28:663, 1979 |
|
Hansen KK, Knopp RH, Soules MR: Lipoprotein-cholesterol levels in infertile women with luteal phase deficiency. Fertil Steril 55:916, 1991 |
|
Fritz MA, Speroff L: The endocrinology of the menstrual cycle: The interaction of folliculogenesis and neuroendocrine mechanisms. Fertil Steril 38:509, 1982 |
|
Minassian SS, Wu CH: Free and protein-bound progesterone during normal and luteal phase defective cycles. Intl J Gynaecol Obstet 43:163, 1993 |
|
Lessey BA, Killam AP, Metzger DA, et al: Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metabol 67:334, 1988 |
|
Bayard F, Damilano S, Robel P, Baulieu E-E: Cytoplasmic and nuclear estradiol and progesterone receptors in human endometrium. J Clin Endocrinol Metabol 46:635, 1978 |
|
Keller DW, Wiest WG, Askin FB, et al: Pseudocorpus luteum insufficiency: A local defect of progesterone action on endometrial stroma. J Clin Endocrinol Metabol 48:127, 1979 |
|
Gautray JP, de Brux J, Tajchner G, et al: Clinical investigation of the menstrual cycle: III. Clinical, endometrial, and endocrine aspects of luteal defect Fertil Steril 34:296, 1981 |
|
Gravanis A, Zorn JR, Tanguy G, et al: The “dysharmonic luteal phase” syndrome: Endometrial progesterone receptor and estradiol dehydrogenase. Fertil Steril 42:730, 1984 |
|
Laatikainen T, Andersson B, Karkkainen J, Wahlstrom T: Progestin receptor levels in endometria with delayed or incomplete secretory changes. Obstet Gynecol 62:592, 1983 |
|
Levy C, Robel P, Gautray JP, et al: Estradiol and progesterone receptors in human endometrium: Normal and abnormal menstrual cycles and early pregnancy. Am J Obstet Gynecol 136:646, 1980 |
|
McRae MA, Blasco L, Lyttle CR: Serum hormones and their receptors in women with normal and inadequate corpus luteum function. Fertil Steril 42:58, 1984 |
|
Saracoglu OF, Aksel S, Yeoman RR, Wiebe RH: Endometrial estradiol and progesterone receptors in patients with luteal phase defects and endometriosis. Fertil Steril 43:851, 1985 |
|
Spirtos NJ, Yurewicz EC, Moghissi KS, et al: Pseudocorpus luteum insufficiency: A study of cytosol progesterone receptors in human endometrium. Obstet Gynecol 65:535, 1985 |
|
McNeely MJ, Soules MR: The diagnosis of luteal phase deficiency: A critical review. Fertil Steril 50:1, 1988 |
|
Hirama Y, Ochiai K: Estrogen and progesterone receptors of the out-of-phase endometrium in female infertile patients. Fertil Steril 63:984, 1995 |
|
Abd-El-Maeboud KH, Eissa S, Kamel AS: Altered endometrial progesterone/oestrogen receptor ratio in luteal phase defect. Dis Markers 13:107, 1997 |
|
Pope WF: Uterine asynchrony: A cause of embryonic loss. Biol Reprod 39:999, 1988 |
|
Hertig AT, Rock J, Adams EC: A description of 34 human ova within the first 17 days of development. Am J Anat 98:435, 1956 |
|
Navot D, Bergh PA, Williams M, et al: An insight into early reproductive processes through the in vivo model of ovum donation. J Clin Endocrinol Metabol 72:408, 1991 |
|
Hodgen GD: Surrogate embryo transfer combined with estrogen-progesterone therapy in monkeys: Implantation, gestation, and delivery without ovaries. JAMA 250:2167, 1983 |
|
Lessey BA, Castelbaum AJ, Sawin SW, Sun J: Integrins as markers of uterine receptivity in women with primary unexplained infertility. Fertil Steril 63:535, 1995 |
|
Lessey BA: Implantation defects in infertile women with endometriosis. Ann N Y Acad Sci 955:265, 2002 |
|
McNatty KP, Sawyers RS, McNeilly AS: A possible role for prolactin in control of steroid secretion by the human Graafian follicle. Nature 250:653, 1974 |
|
Schulz KD, Geiger W, Del Pozo E, Kunzig HJ: Pattern of sexual steroids, prolactin and gonadotropic hormones during prolactin inhibition in normally cycling women. Am J Obstet Gynecol 132:561, 1978 |
|
Andrews WC: Luteal phase defects. Fertil Steril 32:501, 1979 |
|
Scaglia HE, Dron N, Ramos G, et al: Sleep-dependent hyperprolactinemia and corpus luteum pathogenesis. Ricerca Clin Lab 15:159, 1985 |
|
Aisaka K, Yoshida K, Mori H: Analysis of clinical backgrounds and pathogenesis of luteal-phase defect. Horm Res 37(suppl I):41, 1992 |
|
Bahamondes L, Saboya W, Tambascia M, Trevisan M: Galactorrhea, infertility, and short luteal phases in hyperprolactinemic women: Early stage of amenorrhea-galactorrhea? Fertil Steril 32:476, 1979 |
|
Fujimoto VY, Clifton DK, Cohen NL, Soules MR: Variability of serum prolactin and progesterone levels in normal women: The relevance of single hormone measurements in the clinical setting. Obstet Gynecol 76:71, 1990 |
|
Vanrell JA, Balasch J: Prolactin in the evaluation of luteal phase in infertility. Fertil Steril 39:30, 1983 |
|
Soules MR, Bremner WJ, Steiner RA, Clifton DK: Prolactin secretion and corpus luteum function in women with luteal phase deficiency. J Cin Endocrinol Metabol 72:986, 1991 |
|
Cook CL, Schroeder JA, Yussman MA, Sanfilippo JS: Induction of luteal phase defect with clomiphene citrate. Am J Obstet Gynecol 149:613, 1984 |
|
Bonhoff AJ, Naether OG, Johannisson E: Effects of clomiphene citrate stimulation on endometrial structure in infertile women. Hum Reprod 11:844, 1996 |
|
Garcia J, Jones GS, Wentz AC: The use of clomiphene citrate. Fertil Steri1 28:707, 1977 |
|
Keenan JA, Herbert CM, Bush JR, Wentz AC: Diagnosis and management of out-of-phase endometrial biopsies among patients receiving clomiphene citrate for ovulation induction. Fertil Steril 51:964, 1989 |
|
Hsu CC, Kuo HC, Wang ST, Huang KE: Interference with uterine blood flow by clomiphene citrate in women with unexplained infertility. Obstet Gynecol 86:917, 1995 |
|
Hecht BR, Bardawil WA, Khan-Dawood FS, Dawood MY: Luteal insufficiency: Correlation between endometrial dating and integrated progesterone output in clomiphene citrate-induced cycles. Am J Obstet Gynecol 163:1986, 1990 |
|
Check JH, Wu C-H, Adelson HG: Decreased abortions in HMG-induced pregnancies with prophylactic progesterone therapy. Intl J Fertil 30:45, 1985 |
|
Garcia J, Jones GS, Acosta AA, Wright GL: Corpus luteum function after follicle aspiration for oocyte retrieval. Fertil Steril 36:565, 1981 |
|
Frydman R, Testart J, Giacomini P, et al: Hormonal and histological study of the luteal phase in women following aspiration of the preovulatory follicle. Fertil Steril 38:312, 1982 |
|
Graf MJ, Reyniak JV, Battle-Mutter P, Laufer N: Histologic evaluation of the luteal phase in women following follicle aspiration for oocyte retrieval. Fertil Steril 49:616, 1988 |
|
Daya S: Efficacy of progesterone support in the luteal phase following in-vitro fertilization and embryo transfer meta-analysis of clinical trials. Hum Reprod 3:731, 1988 |
|
Buvat J, Marcolin G, Guittard C, et al: Luteal support after luteinizing hormone-releasing hormone agonist for in vitro fertilization: Superiority of human chorionic gonadotropin over oral progesterone. Fertil Steril 53:490, 1990 |
|
Belaisch-Allart J, DeMouzon J, Lapousterle C, Mayer M: The effect of hCG supplementation after combined GnRH agonist/HMG treatment in an IVF programme. Hum Reprod 5:163, 1990 |
|
Herman A, Ron-El R, Golan A, et al: Pregnancy rate and ovarian hyperstimulation after luteal human chorionic gonadotropin in in vitro fertilization stimulated with gonadotropin-releasing hormone analog and menotropins. Fertil Steril 53:92, 1990 |
|
Smitz J, Devroey P, Faguer B, et al: A prospective randomized comparison of intramuscular or intravaginal natural progesterone as a luteal phase and early pregnancy supplement. Hum Reprod 7:168, 1992 |
|
Soliman S, Daya S, Collins J, Hughes EG: The role of luteal phase support in infertility treatment: A meta-analysis of randomized trials. Fertil Steril 61:1068, 1994 |
|
Wakat D, Sweeney K, Rogol A: Reproductive system function in women cross-country runners. Med Sci Sports Exerc 14:263, 1982 |
|
Dale E, Gerlach DH, Wilhite AL: Menstrual dysfunction in distance runners. Obstet Gynecol 54:47, 1979 |
|
Beitins IZ, McArthur IW, Turnbull BA, et al: Exercise induces two types of human luteal dysfunction: Confirmation by urinary free progesterone. J Clin Endocrinol Metabol 72:1350, 1991 |
|
Ellison PT, Lager C: Moderate recreational running is associated with lowered salivary progesterone profiles in women. Am J Obstet Gynecol 154:1000, 1986 |
|
Loucks AB, Mortola JF, Girton L, Yen SSC: Alterations in the hypothalamic-pituitary-ovarian and the hypothalamic-pituitary-adrenal axes in athletic women. J Clin Endocrinol Metabol 68:402, 1989 |
|
Prior JC, Vigna YM: Ovulation disturbances and exercise training. In Pitkin RM (ed): Clinical Obstetrics and Gynecology. Vol 34:pp 180, 190 Philadelphia, JB Lippincott, 1991 |
|
De Souza MJ, Miller BE, Loucks AB, et al: High frequency of luteal phase deficiency and anovulation in recreational women runners: Blunted elevation in follicle-stimulating hormone observed during luteal-follicular transition. J Clin Endocrinol Metab 83:4220, 1998 |
|
Laatikainen TJ: Corticotropin-releasing hormone and opioid peptides in reproduction and stress. Ann Med 23:489, 1991 |
|
Prior JC: Physical exercise and the neuroendocrine control of reproduction. Baillieres Clin Endocrinol Metab 1:299, 1987 |
|
Quinton ND, Laird SM, Okon MA, et al: Serum leptin levels during the menstrual cycle of healthy fertile women. Br J Biomed Sci 56:16, 1999 |
|
Ludwig M, Klein HH, Diedrich K, Ortmann O: Serum leptin concentrations throughout the menstrual cycle. Arch Gynecol Obstet 263:99, 2000 |
|
Thong FS, McLean C, Graham TE: Plasma leptin in female athletes: relationship with body fat, reproductive, nutritional, and endocrine factors. J Appl Physiol 88:2037, 2000 |
|
Paolisso G, Rizzo MR, Mazziotti G, et al: Lack of association between changes in plasma leptin concentration and in food intake during the menstrual cycle. Eur J Clin Invest 29:490, 1999 |
|
Hardie L, Trayhurn P, Abramovich D, Fowler P: Circulating leptin in women: A longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol 47:101, 1997 |
|
Serle E, Aplin JD, Li T-C, et al: Endometrial differentiation in the peri-implantation phase of women with recurrent miscarriage: A morphological and immunohistochemical study. Fertil Steril 62:989, 1994 |
|
Horta JLH, Fernandez JG, de Leon BS, Cortes-Gallegos V: Direct evidence of luteal insufficiency in women with habitual abortion. Obstet Gynecol 49:705, 1977 |
|
Babalioglu R, Varol FG, Ilhan R, et al: Progesterone profiles in luteal-phase defects associated with recurrent spontaneous abortions. J Assist Reprod Genet 13:306, 1996 |
|
Schweiger U: Menstrual function and luteal-phase deficiency in relation to weight changes and dieting. In Pitkin RM (ed): Clinical Obstetrics and Gynecology. Vol 34:pp 191, 197 Philadelphia, JB Lippincott, 1991 |
|
Pirke KM, Schweiger U, Laessle R, et al: Dieting influences the menstrual cycle: Vegetarian versus nonvegetarian diet. Fertil Steril 46:1083, 1986 |
|
Balasch J, Vanrell JA: Mild endometriosis and luteal function. Int J Fertil 30:4, 1985 |
|
Wentz AC: Luteal phase inadequacy. In Sciarra JJ (ed): Gynecology and Obstetrics. Vol 56:pp 1, 10 Philadelphia, JB Lippincott, 1991 |
|
Noyes RW, Hertig AT, Rock JP: Dating the endometrial biopsy. Fertil Steril 1:3, 1950 |
|
Key KD, Kempers RD: Citation classics: Most cited articles from Fertility and Sterility. Fertil Steril 47:910, 1987 |
|
Noyes RW, Haman JO: Accuracy of endometrial dating: Correlation of endometrial dating with basal body temperature and menses. Fertil Steril 4:504, 1953 |
|
Cummings DC, Honore LH, Scott JZ, Williams KP: The late luteal phase in infertile women: Comparison of simultaneous endometrial biopsy and progesterone levels. Fertil Steril 43:715, 1985 |
|
Huang KE: The primary treatment of luteal phase inadequacy: Progesterone versus clomiphene citrate. Am J Obstet Gynecol 155:824, 1986 |
|
Rosenfeld DL, Chudow S, Bronson RA: Diagnosis of luteal phase inadequacy. Obstet Gynecol 56:193, 1980 |
|
Wentz AC, Kossoy LR, Parker RA: The impact of luteal phase inadequacy in an infertile population. Am J Obstet Gynecol 162:937, 1990 |
|
Li T-C, Rogers AW, Lenton EA, et al: A comparison between two methods of chronological dating of human endometrial biopsies during the luteal phase, and their correlation with histologic dating. Fertil Steril 48:928, 1987 |
|
Li T, Dockery P, Rogers A, Cooke I: How precise is histologic dating of endometrium using the standard dating criteria? Fertil Steril 51:759, 1989 |
|
Gibson M, Badger GJ, Byrn F, et al: Error in histologic dating of secretory endometrium: Variance component analysis. Fertil Steril 56:242, 1991 |
|
Scott RT, Snyder RR, Strickland DM, et al: The effect of interobserver variation in dating endometrial histology on the diagnosis of luteal phase defects. Fertil Steril 50:888, 1988 |
|
Castelbaum AJ, Wheeler J, Coutifaris CB, et al: Timing of the endometrial biopsy may be critical for the accurate diagnosis of luteal phase deficiency. Fertil Steril 61:443, 1994 |
|
Balasch J, Vanrell JA, Creus M, et al: The endometrial biopsy for diagnosis of luteal phase deficiency. Fertil Steril 44:699, 1985 |
|
Batista MC, Cartledge TP, Merino MJ, et al: Midluteal phase endometrial biopsy does not accurately predict luteal function. Fertil Steril 59:294, 1993 |
|
Jordan J, Craig K, Clifton D, Soules M: Luteal phase defect: The sensitivity and specificity of diagnostic methods in common clinical use. Fertil Steril 62:54, 1994 |
|
Andoh K, Mizunuma H, Nakazato Y, et al: Endometrial dating in the conception cycle. Fertil Steril 58:1127, 1992 |
|
Balasch J, Vanrell JA, Marquez M, Gonzalez-Merlo J: Endometrial biopsy inadvertently taken in the cycle of conception. Int J Gynaecol Obstet 22:95, 1984 |
|
Rosenfeld DL, Garcia C-R: Endometrial biopsy in the cycle of conception. Fertil Steril 26:1088, 1975 |
|
Sulewski JM, Ward SP, McGaffic W: Endometrial biopsy during a cycle of conception. Fertil Steril 34:548, 1980 |
|
Wentz AC, Herbert CMI, Maxson WS, et al: Cycle of conception endometrial biopsy. Fertil Steril 46:196, 1986 |
|
Hamilton MP, Fleming R, Coutts JR, et al: Luteal phase deficiency: Ultrasonic and biochemical insights into pathogenesis. Br J Obstet Gynaecol 97:569, 1990 |
|
Wu CH, Minassian SS: The integrated luteal progesterone: An assessment of luteal function. Fertil Steri1 48:937, 1987 |
|
Abraham GE, Maroulis GB, Marshall JR: Evaluation of ovulation and corpus luteum function using measurements of plasma progesterone. Obstet Gynecol 44:522, 1974 |
|
Smith SK, Lenton EA, Landgren B-M, Cooke ID: The short luteal phase and infertility. Br J Obstet Gynaecol 91:1120, 1984 |
|
Downs KA, Gibson M: Basal body temperature graph and the luteal phase defect. Fertil Steril 40:466, 1983 |
|
Geisthovel F, Skubsch U, Zabel G, et al: Ultrasonographic and hormonal studies in physiologic and insufficient menstrual cycles. Fertil Steril 39:277, 1983 |
|
Kalogirou D, Antoniou G, Botsis D, et al: Transvaginal Doppler ultrasound with color flow imaging in the diagnosis of luteal phase defect (LPD). Clin Exp Obstet Gynecol 24:95, 1997 |
|
Kupesic S, Kurjak A, Vujisic S, Petrovic Z: Luteal phase defect: Comparison between Doppler velocimetry, histological and hormonal markers. Ultrasound Obstet Gynecol 9:105, 1997 |
|
Miller MM, Hoffman DI, Creinin M, et al: Comparison of endometrial biopsy and urinary pregnanediol concentration in the diagnosis of luteal phase deficiency. Fertil Steril 54:1008, 1990 |
|
Chatterton RTJ, Haan JN, Jenco JM, Cheesman KL: Radioimmunoassay of pregnanediol concentrations in early morning urine specimens for assessment of luteal function in women. Fertil Steril 37:361, 1982 |
|
O’Rourke MT, Ellison PT: Salivary measurement of episodic progesterone release. Am J Phys Anthropol 81:423, 1990 |
|
Vining RF, McGinley RA: The measurement of hormones in saliva: Possibilities and pitfalls. J Steroid Biochem 27:81, 1987 |
|
Tulppala M, Bjorses UM, Stenman UH, Wahlstrom T: Luteal phase defect in habitual abortion: Progesterone in saliva. Fertil Steril 56:41, 1991 |
|
Joshi SG, Rao R, Henriques EE, et al: Luteal phase concentrations of a progestagen-associated endometrial protein in the serum of cycling women with adequate or inadequate endometrium. J Clin Endocrinol Metabol 63:1247, 1986 |
|
Shoupe D, Mishell DRJ, Lacarra M, et al: Correlation of endometrial maturation with four methods of estimating day of ovulation. Obstet Gynecol 73:88, 1989 |
|
Karamardian LM, Grimes DA: Luteal phase deficiency: Effect of treatment on pregnancy rates. Am J Obstet Gynecol 170:958, 1992 |
|
Balasch J, Vanrell JA, Marquez M, et al: Dehydrogesterone versus vaginal progesterone in the treatment of the endometrial luteal phase deficiency. Fertil Steril 37:751, 1982 |
|
Li TC, Ding SH, Anstie B, et al: Use of human menopausal gonadotropins in the treatment of endometrial defects associated with recurrent miscarriage: preliminary report. Fertil Steril 75:434, 2001 |
|
Katz A, Lancet M, Skornik J, et al: Teratogenicity of progestogens given during the first trimester of pregnancy. Obstet Gynecol 65:775, 1985 |
|
Johansson EDB: Depression of the progesterone levels in women treated with synthetic gestagens after ovulation. Acta Endocrinol 68:779, 1971 |
|
Rock JA, Wentz AC, Cole KA, et al: Fetal malformations following progesterone therapy during pregnancy: A preliminary report. Fertil Steril 44:17, 1985 |
|
Chez RA: Proceedings of the symposium “Progesterone, Progestins, and Fetal Development.” Fertil Steril 30:16, 1978 |
|
Simpson JL: Mutagenicity and teratogenicity of injectable and implantable progestins: Probable lack of effect. In Zatuchni GI (ed): Long-Acting Contraceptive Delivery Systems: Proceedings of an International Workshop. Philadelphia, Harper & Row, 1984 |
|
Dehou MF, Lejeune B, Arijs C, et al: Endometrial morphology in stimulated in vitro fertilization cycles and after steroid replacement therapy in cases of primary ovarian failure. Fertil Steril 48:995, 1987 |
|
Devroey P, Palermo G, Bourgain C, et al: Progesterone administration in patients with absent ovaries. Int J Fertil 34:188, 1989 |
|
Critchley H, Buckley CH, Anderson D: Experience with a “physiological” steroid replacement regimen for the establishment of a receptive endometrium in women with premature ovarian failure. Br J Obstet Gynaecol 97:804, 1990 |
|
Bourgain C, Devroey P, Van Waesberghe L, et al: Effects of natural progesterone on the morphology of the endometrium in patients with primary ovarian failure. Hum Reprod 5:537, 1990 |
|
Davies MC, Anderson MC, Mason BA, et al: Oocyte donation: The role of endometrial receptivity. Hum Reprod 5:862, 1990 |
|
Sauer MV, Stein AL, Paulson RJ, et al: Endometrial responses to various hormone replacement regimens in ovarian failure patients preparing for embryo donation. Int J Gynecol Obstet 35:61, 1991 |
|
Miles R, Paulson R, Lobo R, et al: Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: A comparative study. Fertil Steril 62:485, 1994 |
|
Gibbons W, Toner J, Hamacher P, et al: Experience with a novel vaginal progesterone preparation in a donor oocyte program. Fertil Steril 69:96, 1998 |
|
Tavaniotou A, Smitz J, Bourgain C, Devroey P: Comparison between different routes of progesterone administration as luteal phase support in infertility treatments. Hum Reprod Update 6:139, 2000 |
|
Simon JA, Robinson DE, Andrews MC, et al: The absorption of oral micronized progesterone: The effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertil Steril 60:26, 1993 |
|
Maxson WS, Hargrove JT: Bioavailability of oral micronized progesterone. Fertil Steril 44:622, 1985 |
|
Padwick M, Endacott J, Matson C, et al: Absorption and metabolism of oral progesterone when administered twice daily. Fertil Steril 46:402, 1986 |
|
Norman T, Morse C, Dennerstein L: Comparative bioavailability of orally and vaginally administered progesterone. Fertil Steril 56:1034, 1991 |
|
Nahoul K, deZiegler D: Validity of serum progesterone levels after oral progesterone (letter). Fertil Steril 61:790, 1994 |
|
Licciardi FL, Kwiatowski A, Noyes NL, et al: Oral versus intramuscular progesterone for in vitro fertilization: A prospective randomized study. Fertil Steril 71:614, 1999 |
|
Kimzey LM, Gumowski J, Merriam GR, et al: Absorption of micronized progesterone from a nonliquefying vaginal cream. Fertil Steril 56:995, 1991 |
|
Archer D, Fahy G, Viniegra-Sibal A, et al: Initial and steady-state pharmacokinetics of a vaginally administered formulation of progesterone. Am J Obstet Gynecol 173:471, 1995 |
|
Pouly JL, Bassil S, Frydman R, et al: Luteal support after in-vitro fertilization: Crinone 8%, a sustained release vaginal progesterone gel, versus Utrogestan, an oral micronized progesterone. Hum Reprod 11:2085, 1996 |
|
Nillius SJ, Johansson EDB: Plasma levels of progesterone after vaginal, rectal, or intramuscular administration of progesterone. Am J Obstet Gynecol 110:470, 1971 |
|
Miles RA, Paulson RJ, Lobo RA, et al: Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: A comparative study. Fertil Steril 62:485, 1994 |
|
Wang HS, Soong YK: Transvaginal progesterone supplementation increases serum insulin-like growth factor-binding protein-1 levels. Gynecol Endocrinol 10:349, 1996 |
|
Jobanputra K, Toner JP, Denoncourt R, Gibbons WE: Crinone 8% (90 mg) given once daily for progesterone replacement therapy in donor egg cycles. Fertil Steril 72:980, 1999 |
|
Rizk D, Smitz J: Ovarian hyperstimulation syndrome after superovulation using GnRH agonists for IVF and related procedures. Hum Reprod 7:320, 1992 |
|
Forman RG, Frydman R, Egan D, et al: Severe ovarian hyperstimulation syndrome using agonists of gonadotrophin-releasing hormone for in vitro fertilization: A European series and proposals for prevention. Fertil Steril 53:502, 1990 |
|
Loucopoulos A, Ferin M: The treatment of luteal phase defects with pulsatile infusion of gonadotropin-releasing hormone. Fertil Steril 48:933, 1987 |
|
Gautray JP, Jolivet A, Goldenberg F, et al: Clinical investigation of the menstrual cycle: II. Neuroendocrine investigation and therapy of the inadequate luteal phase Fertil Steril 29:275, 1978 |
|
Daly DC, Waiters CA, Soto-Albors CE, Riddick DH: Endometrial biopsy during treatment of luteal phase defects is predictive of therapeutic outcome. Fertil Steril 40:305, 1983 |
|
Murray DL, Reich L, Adashi EY: Oral clomiphene citrate and vaginal progesterone suppositories in the treatment of luteal phase dysfunction: A comparative study. Fertil Steril 51:35, 1989 |
|
Tho PT, Byrd JR, McDonough PG: Etiologies and subsequent reproductive performance of 100 couples with recurrent abortion. Fertil Steril 32:389, 1979 |
|
Daya S: Efficacy of progesterone support for pregnancy in women with recurrent miscarriage: A meta-analysis of controlled trials. Br J Obstet Gynaecol 96:275, 1989 |
|
Csapo AI, Pulkkinen MO, Ruttner B: The significance of the human corpus luteum in pregnancy maintenance: I. Preliminary studies Am J Obstet Gynecol 112:1061, 1972 |
|
Csapo AI, Pulkkinen MO, Wiest WG: Effect of lutectomy and early progesterone replacement therapy in early pregnant patients. Am J Obstet Gynecol 115:759, 1973 |
|
Bergus GR: When is a test positive? The use of decision analysis to optimize test interpretation Fam Med 25:656, 1993 |
|
Nettleman MD: Receiver operator characteristic curves. Infect Control Hosp Epidemiol 9:374, 1988 |
|
Swets JA: Measuring the accuracy of diagnostic systems. Science 240:1285, 1988 |